


In 2023, the healthcare industry went through a transformative period that affected most people associated with the sector. This happened because of the introduction of four significant regulatory updates that impacted a range of stakeholders.
This article reviews the nuances of these regulations, starting with the FDA’s new guidance on Off-The-Shelf Software in Medical Devices. This will help you understand how this guidance mandates risk assessments and enhanced software testing for your digital health application.
The discussion then shifts to the reinforced GMP-validation requirements for medical device software, that provide additional scrutiny on manufacturers and have negative effects on small healthcare businesses.
The article also examines the EU’s revamped GMP Annex 1, focusing on the manufacturing of sterile medicinal products and its alignment with global standards.
Finally, it highlights the NIH Data Management and Sharing Policy, emphasizing its role in promoting transparency and collaboration in NIH-funded research.
By reviewing these regulations and learning more about how they have changed certain processes, you can better prepare and plan for 2024 by understanding how to mitigate and address their impact.
FDA 2023 guidance on off-the-shelf software in medical devices

The first regulation worth mentioning as it’s the most recent one to affect digital health software and off-the-shelf software solutions is introduced by the Food and Drug Administration. In August 2023, the FDA released new guidance on the use of Off-The-Shelf Software (OTSS) in medical devices.
This guidance addresses the integration of OTSS by focusing on ways to reduce the risk to the patient, improve the testing of the software and the methods used for its development. One of the main issues that come with this integration is that it presents various challenges in terms of the applicability of the guidance to some software types, depending on their goal. (2)
To better understand the importance of these limitations, you need to look at the impact that the guidance has had on the separate stakeholder since its release. The biggest changes can be noticed with device manufacturers, software developers, healthcare providers and the patients that benefit from their services.
Medical device manufacturers
The first stakeholder that you need to pay attention to are medical device manufacturers who are directly impacted by the FDA’s regulation. That is because they are now required to ensure that the OTSS used in their devices is compliant with the guidelines. This regulation involves a comprehensive approach to risk mitigation and particularly applies for high-risk OTSS.
Manufacturers must therefore document software configurations, conduct rigorous testing, and manage any changes to avoid unacceptable risks. An example for how each manufacturer can follow it, includes validating the configuration for high-risk OTSS and tracking any patches provided by the OTSS manufacturer.
Software developers and vendors
The next group that is heavily affected by the guidelines are software developers and vendors. Because of the new changes they face heightened responsibilities when creating OTSS for medical devices. They must be aware of the risk levels associated with their software to ensure compliance with FDA standards.
The announcement of the guidelines by the FDA create new difficulties for high-risk OTSS. They include the demand for extensive documentation and rigorous testing since from August 2023 they are now mandatory. The real challenge for developers and vendors lies in providing test plans and reports. This is especially tricky for open-source software, where having such documentation may not be readily available for everyone.
Healthcare providers
As end-users of medical devices that incorporate OTSS, healthcare providers must also be well-informed about the software’s functionalities and limitations. This knowledge is crucial to ensure patient safety and compliance with the FDA guidelines.
For instance, healthcare providers need to understand the specific actions to be taken for high-risk OTSS to prevent unacceptable risks, including adhering to specified user actions and software configurations.
Patients
The last stakeholder that you need to pay attention to when it comes to understanding the FDA’s guidelines are the patients. Although they are indirectly affected, the safety and effectiveness of medical devices incorporating OTSS directly impact their health outcomes.
For example, a patient relying on a medical device with low-risk OTSS can expect minimal safety concerns, whereas a device with high-risk OTSS requires more stringent risk assessments and controls. The new guidelines allow for this distinction to exist and for further scrutiny over the whole approval process of medical devices.
FDA 2023 focus on product software validation for GMP-validated software
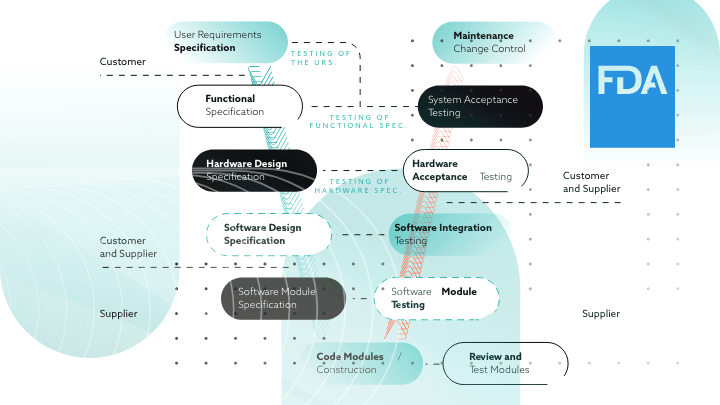
A continuous trend that has characterized the FDA’s goals during 2023 has been its increased focus on product software validation for GMP-validated software. Throughout the year it has made significant changes to its GMP-validation process, especially for medical device and non-product software used in regulated life sciences applications.
This increased focus under 21 CFR Part 820.30, was evidenced by warning letters sent to companies like iRhythm and Zyto. These letters have highlighted the FDA’s growing focus on medical device startups. That’s why it’s important to take a closer look at the impact that this second regulation has had on the different stakeholders to change the way they operate to be “GMP-validated”.
Medical device manufacturers
On top of being affected by the above-mentioned new FDA guidelines, medical device manufacturers are now also under increased scrutiny to ensure proper GMP-validation of their medical device software. The warning letters to iRhythm and Zyto focused on issues like not validating medical device software properly and not verifying infrastructure software used in manufacturing quality processes.
These issues emphasize the need for medical device manufacturers to thoroughly validate both product and non-product software. This information is crucial and has a large impact across the sector, given the fact that over 20% of medical device recalls are GMP-software-related.
Small healthcare and life sciences companies
Small healthcare and life sciences companies have also been affected by this update in the validation process. The pressure has been particularly evident in startups that are being audited a lot more frequently. This trend signifies the FDA’s intent to ensure that even smaller companies adhere to software validation standards.
The complex process of FDA software validation poses a significant challenge for many. This largely happens because the process lacks specific instructions from the FDA. Unless you receive specific guidance for your company by experts, getting your medical device GMP-validated can be difficult. You and your company must demonstrate that your software can accurately and consistently produce results that meet predetermined guidelines for compliance and quality management.
Software development and IT infrastructure teams
On a different note, IT infrastructure teams within regulated companies face the challenge of validating their network infrastructure that supports computerized systems. The GMP-validation process, especially after the new changes, includes demonstrating the adequacy of both hardware and software in a platform system. Along with the application software the validation update set by the FDA also requires that ancillary equipment, people, and procedures are fit for their intended use.
The responsibility of maintaining the software GMP-validation status of the system falls on both the business and system owners. The entire network infrastructure, including components like servers, routers, and network software, needs to be qualified and GMP-validated. This process is essential for ensuring that the infrastructure remains in compliance with the new FDA GMP-validation standards.
EU GMP Guidelines – Annex 1 “Manufacture of Sterile Medicinal Products”

The third regulation under review in this article is connected to the European Commission that revised its Good Manufacturing Practices (GMP) Annex 1 by focusing on sterile drug products. This extensive revision, taking effect primarily in August 2023 and addresses contamination control and sterile processing technologies.
It includes changes to risk management, personnel hygiene, environmental monitoring, and sterility assurance rules. With these changes the European Commission aims to align with WHO and PIC/S standards and the FDA’s 2004 guidance on sterile drug products. The changes aim to harmonize sterile drug manufacturing principles and introduce new sections on pharmaceutical quality systems.
Pharmaceutical companies manufacturing sterile products
The pharmaceutical industry has undergone significant changes since the last GMP revision, especially with the rise of novel biologic therapies. The new GMP Annex 1 differs as it mandates additional safety and contamination control measures.
Another key change is the requirement for Pre-Use Post-Sterilisation Integrity Testing (PUPSIT) and the implementation of a documented CCS across facilities, by focusing on all aspects of contamination control. Many biopharma manufacturers have already been preparing for this change by integrating PUPSIT into their production lines and adopting innovative sterile manufacturing technologies.
Manufacturers developing new processes and systems
Manufacturers developing new compliant manufacturing processes or updating their systems are incorporating standardized solutions to ensure GMP-compliance.
Meanwhile, pre-validated GMP-compliant single-use assemblies are being used as a solution to meet the new manufacturing needs. These plug-and-play systems accelerate the setup of processes, thereby increasing productivity and mitigating risk for users and patients.
Clinical trials and marketing authorization applicants
Finally, when it comes to clinical trials, the revised GMP Annex 1 could lead to longer lead times for the development and approval of new drug products. This will largely be a result of the new requirements on the manufacture of sterile medicinal products. The modifications in existing pharmaceutical manufacturing processes may affect ongoing clinical trials or marketing GMP-authorization applications.
These modifications may potentially require trials to be paused until changes are implemented and GMP-validated. The emphasis on risk management and environmental monitoring in the revised guidelines also means more extensive testing and documentation. This is exactly what could lead to longer approval times for marketing authorization applications.
2023 NIH data management and sharing policy

The last but not least of the important regulations is the NIH Data Management and Sharing (DMS) policy that became effective on January 25, 2023. The policy was established to promote the sharing of scientific data from NIH-funded research. It aims to accelerate biomedical research discovery by enabling the validation of research results. It does that by enhancing accessibility to high-value datasets and promoting data reuse for future studies.
The NIH policy also requires investigators and institutions to plan and budget for managing and sharing data, submit a DMS plan for review when applying for funding, and comply with the approved plan. The policy is a significant step towards maximizing public access to research results funded by NIH.
NIH-funded researchers
The 2023 DMS Policy represents a significant change from the previous NIH Data Sharing Policy implemented in 2003. It removes the previous budget threshold of $500,000 direct cost per year and now requires any NIH-funded researcher to provide a detailed DMS Plan.
This plan must now outline the data to be shared, the availability timeline, preservation locations, and any limitations on data sharing. Researchers are expected to integrate these plans into their grant planning and administration processes, ensuring transparency and accessibility of research data.
Institutions conducting NIH-funded research
The new policy places a substantial responsibility on institutions conducting NIH-funded research. These institutions must oversee the implementation of DMS plans and ensure compliance with NIH guidelines.
Dr. Lawrence Tabak, performing the duties of NIH director, noted that the policy strikes a balance between reasonable expectations for data sharing and flexibility for diverse data types and circumstances. It establishes data sharing as a fundamental component of the research process and enhances the public’s access to results from NIH-funded research.
Conclusion
These regulatory changes show you how the healthcare landscape has evolved in 2023. The nuances of the FDA’s 2023 guidance on Off-The-Shelf Software in Medical Devices emphasize the increased focus on risk management and testing for digital health software. At the same time, the significant revisions in the EU’s GMP Annex 1 process are related to sterile drug production and contamination control.
From digital health software’s premarket submission requirements to the validation of GMP-validated software and the management of data in clinical research, each regulation presents unique challenges and opportunities. By better understanding them you can make sure you stay compliant with each of the new guidances and regulations. Staying updated can be a challenge, so make sure to book a call with one of our specialists if you feel like you need extra guidance.
Delyan Stoyanov, co-founder of XTATIC Health, combines his background in medical education with specialized expertise in medical copywriting and marketing.
link to the author’s linkedin profile



