


In the last decade, healthcare technology has advanced significantly. Many fields, such as cardiology, home care, and dentistry, have developed their treatment techniques and received new medical devices.
However, the greatest development in the field was in digital health technology. Many companies started developing digital health solutions. They created software as a medical product or medical devices connected to the Internet, thus establishing a digital medical device industry. All of this meant that large amounts of personal data began to be received and processed over the Internet.
Naturally, this put data privacy and safety at the forefront of lawmakers’ attention. For this reason, virtually every state is developing its regulatory systems. Specifically, the USA and the EU have the greatest influence in the field with the Health Insurance Portability and Accountability Act (HIPAA) and the General Data Protection Regulation (GDPR), respectively.
These frameworks establish comprehensive standards for digital safety and health compliance across their respective jurisdictions.
This article will compare the European and American approaches to digital health regulation. We’ll examine the legal frameworks governing health data, the agencies enforcing regulatory standards, and the specific regulations for medical devices. Finally, we’ll provide practical tips for businesses operating across both markets.
Digital health compliance in Europe
Europe’s approach to digital health centers around protecting individuals’ data while promoting innovation. The General Data Protection Regulation (GDPR) serves as the cornerstone of this framework, even though it wasn’t developed specifically for healthcare.
The GDPR regulates any situation where a company processes personally identifiable information (PII) of an individual. In the context of healthcare, PII covers not just medical data but any sensitive data that can directly or indirectly identify a person – including names, addresses, telephone numbers, and even online identifiers.
Under European regulations, data generated must be used for legitimate purposes. Simply keeping information “just in case” isn’t considered valid. Companies need specific reasons for data processing, such as user consent, contractual obligations, or legal requirements, as outlined in Article 6 of the GDPR.
The European system puts significant emphasis on transparency and individual rights. Users must be aware of how their data is handled and have meaningful control over its processing. A feature unique to European legislation is the “right to be forgotten,” which allows individuals to request complete deletion of their personal data from an organization’s databases.
Data breach notification requirements in Europe are particularly strict. When personal data is compromised, organizations must report the breach to relevant authorities within 72 hours. This short window creates significant compliance challenges for digital health providers.
Violations can result in substantial penalties. Organizations that fail to comply with GDPR face sanctions of up to 4% of their annual global revenue or €20 million, whichever is higher. For example, DEDALUS BIOLOGIE, which distributes software for laboratories, received a €1.5 million fine after a data leak exposed the personal information of 500,000 individuals.
To enforce these standards, each EU member state has established independent supervisory authorities. These national bodies coordinate through the European Data Protection Board, ensuring consistent application of data protection rules across the European market.
For digital health companies, these rigorous standards create both challenges and opportunities. While compliance requires significant investment, the resulting trust and standardization can become competitive advantages in the growing European digital health market.
Digital health compliance in the USA
The United States takes a different approach to digital health regulation compared to Europe. Rather than using a single comprehensive framework, the US relies on sector-specific regulations. The Health Insurance Portability and Accountability Act (HIPAA) serves as the primary law governing health information privacy.
HIPAA is more targeted than GDPR, focusing specifically on protected health information (PHI). It applies to each covered entity – healthcare providers, health plans, and healthcare clearinghouses – as well as their business associates. Unlike the European approach, organizations outside these categories may not be subject to these specific health data protections.
The American system provides healthcare providers with more flexibility in data sharing. PHI can be disclosed without patient consent for treatment, payment, and healthcare operations. “Treatment” covers providing healthcare services. “Payment” relates to billing and insurance matters. “Healthcare operations” include administrative, legal, and quality improvement activities that don’t directly treat patients.
Data breach notification requirements in the US allow more time for response than in Europe. Healthcare providers must notify the Department of Health and Human Services Office for Civil Rights (OCR) when PHI is breached. For incidents affecting more than 500 individuals, notifications must be made within 60 days. Smaller breaches can be reported annually by March 1st of the following year.
Penalties for violations depend on the level of negligence involved. Fines range from $100 to $50,000 per violation when an organization couldn’t reasonably have known about the violation. Cases involving willful neglect can result in fines between $10,000 and $50,000 per violation, with possible criminal charges.
The maximum annual penalty for identical violations is $1.5 million. A notable example is Anthem Inc., which faced a $115 million class-action lawsuit after hackers accessed the data of 79 million people.
US digital health landscape
The US digital health landscape emphasizes innovation, particularly in the mobile health and IT sectors.
Multiple agencies coordinate the regulation of new wireless medical devices, device software functions, and health applications:
- The Food and Drug Administration (FDA) oversees equipment intended for the treatment, prevention, or diagnosis of diseases.
- The Federal Communications Commission (FCC) manages equipment using radio frequencies.
- The Office of the National Coordinator for Health Information Technology (ONC) works on interoperable health information technology.
This distributed approach allows for specialized oversight. The FDA has approved various mobile applications, including Mobile MIM, which allows doctors to view medical images, and Mobisante, a smartphone ultrasound system.
Beyond these agencies, the United States continues to develop its digital health policy through various proposed regulatory frameworks. The Federal Trade Commission Act also plays a role in regulating digital health products by prohibiting deceptive trade practices in how companies market their solutions.
As connected devices generate data and artificial intelligence becomes more prevalent in healthcare, regulatory authorities are establishing clearer pathways for enforcement discretion. This is particularly important for pharmaceutical companies developing digital therapeutics that require clinical investigation and evaluation.
These evolving frameworks reflect the broad range of health-related software intended for medical purposes, requiring robust privacy and security measures across the digital transformation of healthcare.
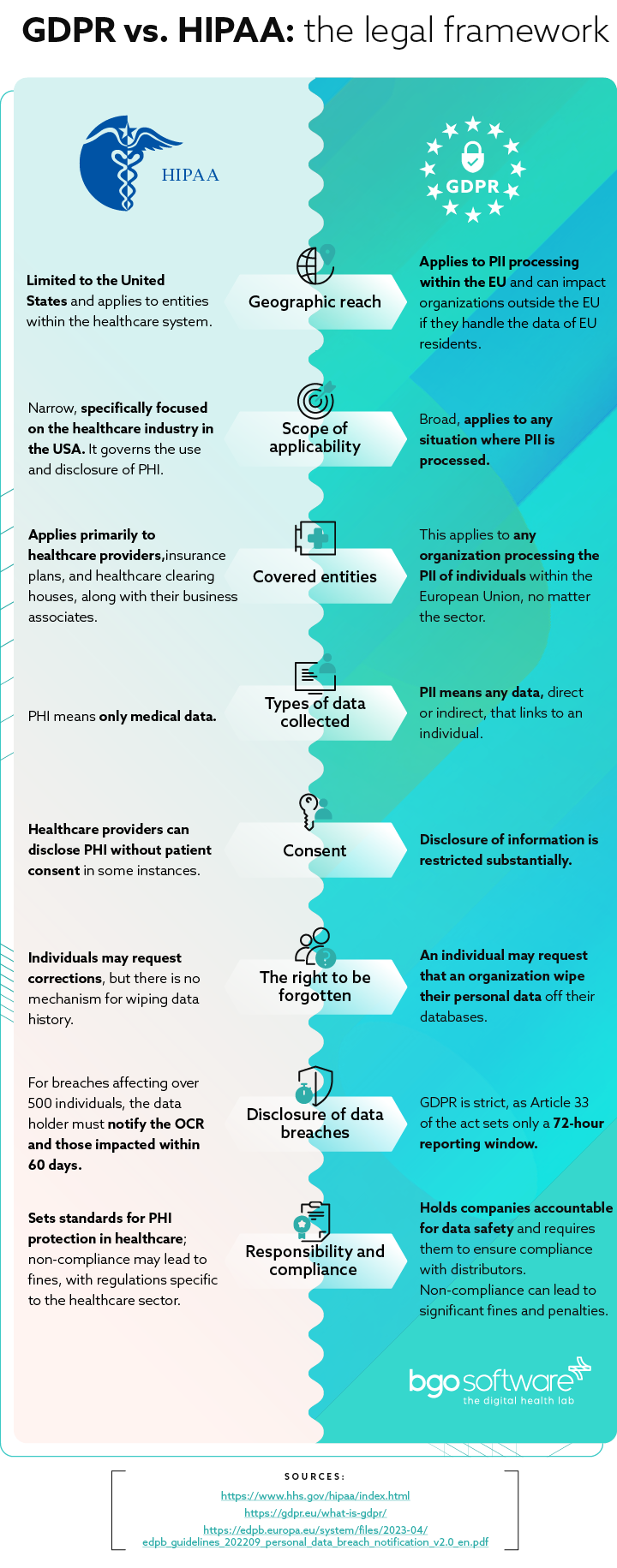
The legal framework: GDPR vs. HIPAA

The fundamental differences between GDPR and HIPAA become clear when examining specific aspects.
Types of data collected
Under US law, PHI means only medical data – meaning the condition of the patient, chronic diseases, etc. are protected.
In the EU, PII means any data, direct or indirect, that links to an individual – name, address, telephone number, skin color, etc. However, indirect information can also include online history, preferences, health status, and more.
Consent
Under HIPAA, healthcare providers can disclose PHI without patient consent in some instances. A disclosure with another medical provider is permissible for treatment, payment purposes, and healthcare operations. “Treatment” means providing healthcare in general. “Payment” is related to billing patients for treatment, obtaining reimbursement, and handling insurance. “Healthcare operations” are any legal, administrative, or quality improvement actions that do not treat the patient itself.
However, under GDPR, such disclosure of patient data is restricted substantially. Only information related to direct patient care or “treatment” can be shared without explicit consent. Anything outside of that scope requires explicit consent. This includes communication and marketing activities between the care provider and data subject – the individual must expressly consent to communications through phone, e-mail, etc.
The right to be forgotten
A feature unique to European legislation is the increased control over personal information records. An individual may request that an organization wipe their personal data off their databases. This includes wiping such data from associates or affiliates. This means that an organization needs to be able to track where PII is stored and have access to the database.
HIPAA permits patients to view their own medical records and other health information. Furthermore, individuals may request corrections, but there is no mechanism for wiping data history.
Disclosure of data breaches
When the PHI of a patient is breached, healthcare providers must notify the Department of Health and Human Services Office for Civil Rights (OCR). If more than 500 individuals are affected by such a breach, the data holder must notify the OCR and all affected individuals within 60 days. If a smaller number of people are affected, such reporting must be done by the last day of reporting each year – or they must report the incident by the 1st of March.
Yet, when it comes to the EU, GDPR is drastically stricter, as Article 34 of the act sets only a 72-hour disclosure period.
Violations
HIPAA violations vary considerably, depending on each particular case and the severity of the violations. They are based on whether a healthcare provider could not have known that his actions would result in a violation or whether he was negligent. In the first case, the fines range from $100 to $50,000. In the second, they range from $10,000 to $50,000, with possible criminal charges and jail time. Lastly, the penalty can be up to $1.5 million a year for violating an identical provision.
GDPR violations are dealt with in a simpler way. Organizations that violate the law face sanctions up to 4% of their income in the last 12 months or 20 million euros, depending on which is higher.
To issue such fines, each state needs competent authorities that monitor compliance with HIPAA and GDPR. For this reason, the FDA and EMA both monitor the healthcare field, from the pharmaceutical industry’s technology and drugs affecting human health to diagnostic and interventional radiology and medical devices worldwide.
Regulatory standards: the role of EMA and FDA in digital health

Beginning with an explanation of the organizations. The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) both operate under very similar regulatory frameworks and share more differences than HIPAA and GDPR.
They are the competent authorities responsible for testing the safety and efficiency of a medical device, protecting and advocating for public health, and giving people health and regulatory information. There are only three main differences between the two agencies:
Structure
The FDA operates and oversees the USA exclusively. Due to this, it is centralized and oversees the drug approval process with its own staff.
The EMA, on the other hand, overviews the same process in many European countries. Due to this, centralization has not yet occurred, which leads to some complications. The assessment of new drugs and devices is done independently by each country, and the agency brings resources from more than 40 national competent authorities and more than 4500 experts. After the assessment, the EMA sends an opinion to the European Commission, which is either approved or denied.
Phases of approval
In both agencies, there are three stages that define the regulatory pathway – preclinical testing, clinical trials, and a final approval procedure. In the USA, an application is filed with the FDA for drugs that appear safe in the preclinical phase.
In the EU, an application is filed for receiving a marketing authorization license, which is valid in every EU state plus Iceland, Lichtenstein, and Norway. Most but not all products in the EU must follow the centralized authorization procedure, but some products may still be authorized through national decentralized procedures.
Differences in testing
The testing process itself is quite similar for the two agencies, with only a slight twist. The FDA investigates new drugs when compared to placebos and increasingly considers real-world evidence, while the EU compares the new drugs with old medications. However, this is not always the case, as the EMA does also incorporate placebo and active treatment as controls when possible.
Yet, the trend is toward standardization of the approval mechanisms. Specifically, the FDA and the EMA already have the same application form for rare diseases. This common framework allows companies to apply in both jurisdictions at the same time.
That being said, while the data protection aspect of digital health regulation is relatively similar, there are substantial differences on the hardware front.
Medical device regulations in the EU and USA

When speaking about digital health, medical devices are usually not the first thing a person thinks about. However, many hardware medical devices are connected to the Internet.
For example, the Internet of Things (IoT) is a line of devices – sensors, software, and other digital health technologies. These are created for exchanging data with other devices over the Internet. Remote patient monitoring is a key application in this field. Remote heart monitoring devices track patients from their homes and submit the data to healthcare professionals.
In this sense, remote heart monitoring devices could be used to track patients from their homes and submit the data gathered to healthcare professionals through the Internet. In that sense, the regulation of medical devices has just as much importance for digital healthcare.
Regulation in the European Union
Particularly in Europe, lawmakers have recognized several emerging challenges – the aging population, rising expectations of patients, and the migration of patients and health professionals.
To that end, the focus has been on branches like E-health, M-health, and genomics – all in an attempt to switch their approach from treating underlying conditions to the prevention of illnesses in the first place.
The EU defines medical devices as products or equipment intended for a medical purpose and regulates them under the New Approach (NA) directives. Under the directive, products placed on the EU market and benefiting from the free movement rules are covered by the legislative harmonization standards.
These standards are technical specifications that ensure the safety of the product. The key part is that these standards are not mandatory, and the manufacturer can apply other national standards. However, approved devices under the harmonized standards of the NA receive a presumption of conformity with the required standards in every EU country.
The assessment of the product itself is based on the risk levels and the intended use of the device.
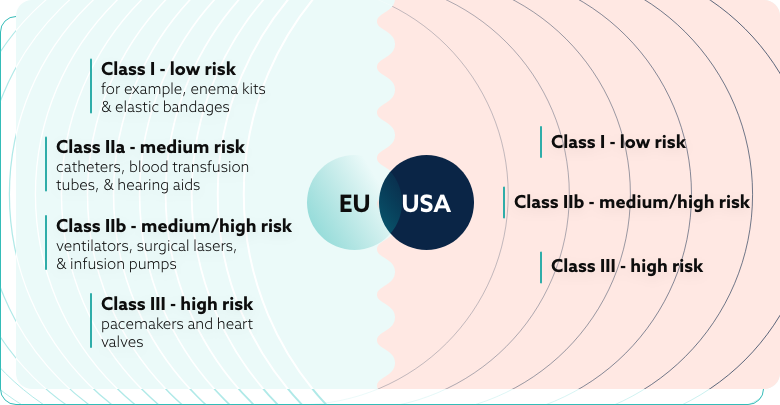
The EU medical device classification system includes four distinct categories:
- Class I – low risk – for example, enema kits and elastic bandages;
- Class IIa – medium risk – catheters, blood transfusion tubes, and hearing aids;
- Class IIb – medium/high risk – ventilators, surgical lasers, and infusion pumps;
- Class III – high risk – pacemakers and heart valves.
These strict regulations are created to ensure that the medical device can accomplish its intended use without compromising the condition or safety of the user. Any risk that is found is weighed against the possible benefit to the patient, ensuring the product is likely to produce more good than harm.
Regulation in the United States of America
The USA market is characterized by the development of the mobile and health IT sectors. Lawmakers are undergoing an effort to coordinate their approach towards the new wireless medical devices, mobile apps, and other digital health devices.
The enforcement process is significantly more centralized, with the FDA, the Federal Communications Commission (FCC), and the Office of the National Coordinator for Health Information Technology (ONC) executing the main regulatory actions.
The FCC mainly handles international communication by telephone, radio, television, internet, etc. It also oversees the authorization of equipment using radio frequencies. Lastly, it examines equipment emitting radio frequency energy that could cause interference with other systems.
The FDA, on the other hand, extends its oversight to equipment intended for the treatment, prevention, or diagnosis of diseases. FDA’s oversight includes various digital health technologies.
One example of such authorization is Mobile MIM, a program whose software code is capable of “allowing doctors to view and assess medical images that have been approved by the FDA”.
Another example is Mobisante, an app that is a “mobile ultrasound imaging system that will cost between $7,000 and $8,000 in full” and displays the ultrasound on a smartphone.
The two agencies are coordinating their efforts and have even signed a memorandum to share information on device marketing authorizations and consult each other on the development of standards for mobile devices and digital health IT.
Lastly, the ONC is charged with the development and implementation of interoperable information technology. Some of its obligations might be to ensure rigorous regulation of certification programs for health IT or to develop standards for the certification of said programs.
Medical devices
In the US, medical devices are defined as “instruments or apparatus (including components) intended for use when diagnosing, treating or preventing diseases or medical conditions, or intended to affect the body through non-chemical means.”
Some accessories used for marketing purposes or general use and not strictly for healthcare do not fall under the control of the above-mentioned organizations.
However, as in the EU, the intended use of the device is taken into account to determine if it is a medical device.
Similarly to Europe, the devices are classified based on their risk factors and efficiency:
- Class I – low-risk devices;
- Class II – moderate-risk devices;
- Class III – high-risk devices.
Manufacturers are generally expected to classify their own devices before commercializing them based on risk assessment. However, most of them are still subject to “general controls”, such as device listing and good manufacturing practice requirements and reporting.
Tips for intercontinental businesses
The current regulatory frameworks in Europe and the United States are quite similar when it comes to the protection of personal data in healthcare. The regulatory requirements of both GDPR and HIPAA were not created specifically for healthcare, they are both perfectly applicable in the field.
When it comes to comparing the two laws, the European law is more decentralized due to the nature of the European Union.
Despite this, GDPR is quite stricter than HIPAA in many aspects, such as their policy around sharing information without the consent of the individual, the right to be forgotten, or the fines issued for violations of the act. Yet, the updated regulatory changes from recent years show a trend toward creating equal standards in both laws.
This similarity is not present in their enforcement agencies – the EMA and FDA, however, both still use similar techniques for assessing digital software and medical devices. In fact, both American and European agencies have similar standards for assessing the risk related to each medical product.
For a business, this means that software or a medical device authorized in Europe has a great chance of being ready to begin the approval process in the USA, as the EU standards tend to be stricter. On the flip side, a product may be sent to the European market with relatively few changes.
Of course, there are always exceptions, and when dealing with regulations, even small mistakes can lead to fines. However, working with an IT company focused on medical software could be the key to an easy transition.
Working with such companies means that they will catch small mistakes that are easy to overlook, and instead of paying for experience with fines, a company can pay a smaller fee to a third party to ensure regulatory compliance.
Recommendations
When operating across both markets, consider these additional recommendations:
- Start with developing GDPR-compliant systems if you plan to operate in both markets. Meeting the stricter European standards will typically satisfy American requirements as well.
- Pay special attention to breach notification timelines. Having response protocols ready for Europe’s 72-hour window will keep you compliant in both regions.
- Develop clear data classification systems that identify which information falls under PHI (for HIPAA) and PII (for GDPR) to ensure appropriate protection levels.
- Consider the different consent models when designing user interfaces and data collection processes. European operations require more explicit consent mechanisms.
- For medical devices, understand which risk category your product falls into early in the development process. This knowledge allows you to build appropriate compliance measures from the beginning.
By carefully navigating these regulatory differences, digital health companies can successfully operate across both European and American markets. This approach enables delivering innovations while maintaining compliance with increasingly complex data protection requirements.
Ivan is a Technical Copywriter with extensive experience in the field of medical technology and software development. He specializes in translating complex technical concepts into clear and engaging content tailored for both industry professionals and broader audiences.
link to the author’s linkedin profile



