


Software is no longer a supporting tool in life sciences – it is embedded directly into how products are developed, manufactured, tested, released, and monitored. From batch execution and laboratory workflows to deviation handling and quality decision-making, computerized systems increasingly are the process.
At the same time, many validation programs still reflect assumptions from an earlier era: static systems, infrequent updates, and heavy reliance on scripted testing and documentation. The result has often been a growing gap between regulatory intent and industry practice with validation perceived as slow, costly, and resistant to change.
The FDA’s guidance on Computer Software Assurance (CSA) addresses this gap. CSA is not a deregulation effort, nor a replacement for validation. Instead, it clarifies how manufacturers can apply risk-based, least-burdensome assurance to modern software systems while maintaining compliance, quality, and patient safety.
Key takeaways
-
Computer Software Assurance (CSA) is a risk-based approach recommended by the FDA for establishing and maintaining confidence that software is fit for its intended use.
-
CSA does not replace Computer System Validation (CSV); it clarifies how validation activities may be executed more effectively.
-
CSA applies across Pharma and MedTech environments, including MES, LIMS, QMS, CAPA systems, CPV analytics, and Digital Validation Tools (DVTs).
- CSA aligns with existing regulatory frameworks such as 21 CFR Part 820, Part 11, EU GMP Annex 11, and GAMP 5.
-
The focus shifts from document volume to meaningful assurance, helping organizations keep systems reliable and in a validated state over time.
What is Computer Software Assurance (CSA)?
Computer Software Assurance is a risk-based approach recommended by the FDA for establishing and maintaining confidence that software used in production or quality systems is fit for its intended use.
Rather than prescribing uniform testing and documentation across all systems and functions, CSA evaluates:
-
-
How the software is used
-
The potential impact of failure on product quality or patient safety
-
The level of assurance necessary to manage that risk
The FDA’s position is that applying this approach allows manufacturers to focus quality assurance activities where they matter most, while still meeting existing validation requirements.
CSA does not remove regulatory obligations. It provides guidance on how to fulfill them more effectively.
Why did the FDA introduce CSA?
CSA emerged from a broader realization within the FDA that validation practices were unintentionally discouraging modernization.
Through its Case for Quality initiative, the FDA observed a recurring pattern:
-
Organizations delayed system upgrades
-
Kegacy software remained in use longer than intended
-
Automation and digital tools were underutilized
The underlying reason was not regulatory prohibition, but perceived validation burden especially for systems that change frequently or rely on cloud and configurable platforms.
Key drivers behind CSA include:
-
Technological advancement
The FDA recognizes that automation, robotics, simulation, and advanced analytics can improve quality and reduce human error when implemented responsibly. -
Need for clarity and agility
Manufacturers sought clearer expectations for validating software that evolves iteratively rather than remaining static. -
Focus on real risk
Excessive testing of low-risk functionality does not necessarily improve outcomes. CSA emphasizes assurance proportional to risk.
CSA was developed through collaboration across multiple FDA centers (CDRH, CBER, CDER, OCP, ORA), signaling its relevance across medical devices, biologics, and pharmaceuticals.
The core principle: more testing, less documentation
The FDA Computer Software Assurance approach provides value by fundamentally shifting the focus from administrative tasks to core quality assurance, effectively addressing the regulatory goals.
Under CSA, the effort is reversed from the old CSV mindset:
- 80% of effort is directed toward meaningful, risk-based testing. This significantly enhances patient safety and data integrity by thoroughly verifying the most critical system functions. The focus is on proving the system works reliably.
- Only 20% of effort is dedicated to right-sized, value-adding documentation. This drastic reduction in paperwork leads to accelerated validation and greater operational efficiency, as companies spend less time creating and reviewing unnecessary records.
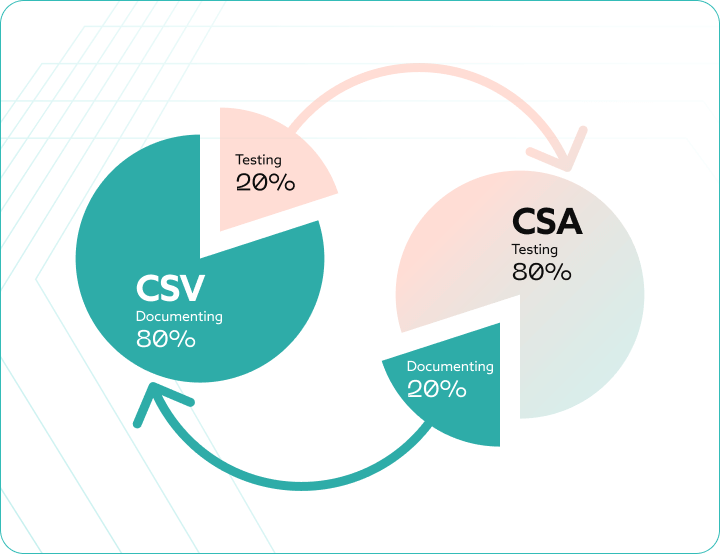
The core value is a practical shift that results in a faster and more effective validation process by prioritizing system quality over the volume of documentation.
What has not changed: validation is still required
One point must be stated clearly:
Computer System Validation remains a regulatory requirement.
Any computerized system used in regulated activities must be demonstrated to be fit for its intended use and maintained in a controlled state throughout its lifecycle.
CSA does not eliminate:
-
Validation planning
-
Testing
-
Documentation
-
Inspection accountability
What changes is how assurance activities are selected, scaled, and justified.
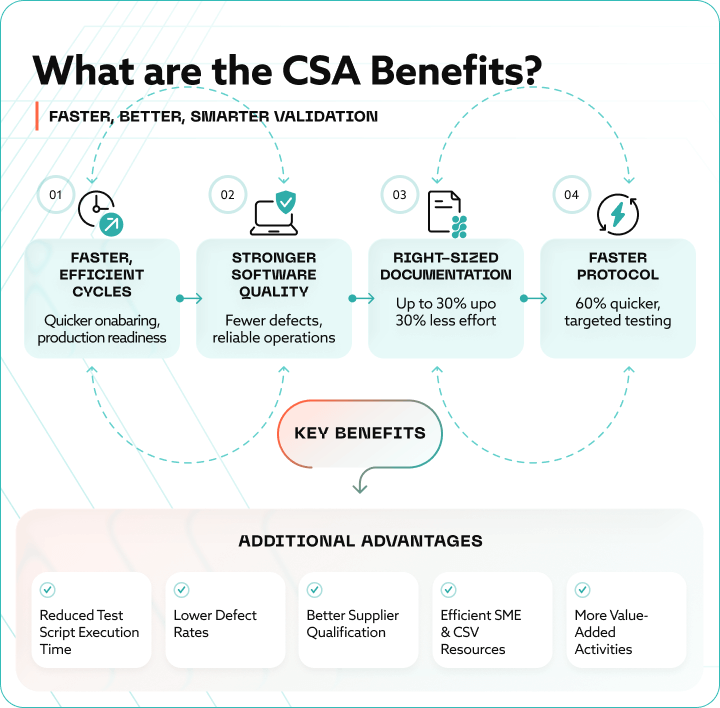
What organizations gain by applying CSA principles
Organizations that adopt CSA-aligned validation practices typically experience several practical benefits:
-
More efficient validation cycles
By avoiding over-testing of low-impact functionality, teams can progress through validation activities more efficiently—especially for configurable or frequently updated systems. -
Improved focus on software quality
Assurance effort shifts toward critical workflows and failure modes, increasing the likelihood that real issues are identified early. -
Reduced documentation overhead without loss of control
CSA encourages fewer, more meaningful records that are easier to review, maintain, and defend during inspections. - Better use of subject matter expertise
SMEs and QA professionals spend less time executing low-value scripts and more time evaluating risk, interpreting results, and ensuring systems remain under control.
CSV and CSA: different roles, same regulatory objective
Computer System Validation defines the regulatory obligation to demonstrate fitness for intended use. CSA does not replace this obligation.
What CSA changes is how validation activities are planned and executed.
Traditional practices often applied uniform testing and documentation across entire systems. CSA introduces explicit risk-based differentiation, allowing assurance effort to scale based on the impact of specific software functions.
CSA should therefore be viewed as a clarification and refinement of validation execution, not a departure from CSV. The regulatory expectation to establish and maintain confidence in system performance remains unchanged.
How CSA fits into Pharma and MedTech environments
CSA fits naturally into the digital backbone of Pharma and MedTech operations.
Core systems such as:
-
Manufacturing Execution Systems (MES)
-
Laboratory Information Management Systems (LIMS)
-
Quality Management Systems (QMS)
-
CAPA modules
all depend on reliable software to maintain product quality and compliance.
Many of these systems are built on commercial, configurable platforms, often classified under GAMP Categories 1 or 3, while still containing configurable or custom components. CSA aligns well with this reality by allowing manufacturers to:
-
Assess risk at the function or configuration level
-
Leverage supplier assurance for standard components
-
Focus internal testing on quality-critical or custom functionality
This approach supports efficient validation while remaining consistent with GAMP 5 principles and quality risk management expectations.
Key FDA principles underlying Computer Software Assurance
The CSA guidance reflects several foundational principles that shape FDA expectations:
-
Risk-driven assurance decisions
Assurance effort should be proportional to the potential impact of failure on patient safety or product quality. -
Critical thinking over mechanical execution
Manufacturers are expected to understand their systems and justify why specific assurance activities are sufficient. -
Appropriate reliance on supplier activities
Where suppliers maintain robust quality systems, their assurance activities may be leveraged rather than duplicated. -
Meaningful records, not excessive documentation
Documentation should support assurance conclusions and inspection transparency, not add complexity without benefit. -
Lifecycle perspective
Validation is not limited to system implementation; maintaining a validated state requires ongoing monitoring and effective change control.

What CSA looks like in practice
CSA does not prescribe specific templates or tools. Instead, it guides decision-making.
In practice, CSA typically involves:
-
Defining the intended use of the software or function
-
Assessing the potential risk of failure
-
Selecting assurance activities appropriate to that risk
-
Maintaining records that support the conclusions reached
High-risk functions may still require structured, scripted testing. Lower-risk functionality may be assessed through exploratory or scenario-based approaches, provided outcomes and rationale are documented.
Is CSA mandatory? Understanding the FDA’s position
CSA is nonbinding guidance, not regulation.
The FDA explicitly states that guidance documents represent the Agency’s current thinking and do not establish legally enforceable requirements. Manufacturers may use alternative approaches if they satisfy applicable statutes and regulations.
CSA supersedes Section 6 of the FDA’s older General Principles of Software Validation guidance but does not replace validation requirements themselves.
CSA supports compliance with:
-
21 CFR Part 820 (Quality System Regulation)
-
21 CFR Part 11 (Electronic Records and Signatures)
-
Related guidance documents
The FDA encourages adoption of CSA principles to help manufacturers keep pace with rapidly evolving technology while maintaining control.
Frequently Asked Questions (FAQ)
What is the main purpose of CSA?
To provide a risk-based approach for establishing and maintaining confidence that software is fit for its intended use.
How does CSA differ from traditional CSV practices?
CSA refines how validation activities are executed by scaling assurance based on risk, rather than applying uniform testing and documentation.
Is CSA mandatory for pharmaceutical companies?
No. CSA is guidance that recommends a method for fulfilling existing validation requirements.
How does CSA align with GAMP 5 and Part 11?
CSA aligns with GAMP 5’s risk-based principles and supports a least-burdensome approach to ensuring systems subject to Part 11 perform as intended.
What are the key benefits of CSA?
Improved focus on quality and patient safety, more efficient validation, and better support for modern digital systems.
Looking ahead: validation that supports innovation and control
Computer Software Assurance reflects a broader regulatory signal: validation should enable, not inhibit, responsible use of modern technology.
By focusing assurance effort on real risk, CSA helps organizations maintain control while adapting to increasingly dynamic software environments.
Manufacturers that approach CSA thoughtfully grounded in risk management, system understanding, and clear justification are better positioned to sustain compliance while continuing to evolve.
References:
[1] American Pharmaceutical Review. (2021, December 6). Understanding FDA’s CSA guidance in the context of current regulations and GAMP® 5. https://www.americanpharmaceuticalreview.com/Featured-Articles/574659-Understanding-FDA-s-CSA-Guidance-in-the-Context-of-Current-Regulations-and-GAMP
[2] U.S. Food and Drug Administration, Center for Devices and Radiological Health, & Center for Biologics Evaluation and Research. (2025, September 24). Computer Software Assurance for Production and Quality System Software: Guidance for industry and Food and Drug Administration staff. U.S. Department of Health and Human Services.
[3] U.S. Food and Drug Administration. (2025). Title 21, Chapter I, Subchapter H, Part 820: Quality System Regulation (Up to date as of October 28, 2025). eCFR.
[4] U.S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), & Center for Veterinary Medicine (CVM). (2018, December). Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry. Food and Drug Administration.
Yoanna is a Technical Copywriter with a keen interest in healthcare innovations and medicine. She is dedicated to crafting clear and engaging content that highlights the latest advancements and trends in the medical field.
link to the author’s linkedin profile



