


For years, the regulated industry has often followed the maxim: “If it’s documented, it’s compliant.” But is the goal truly to validate paperwork, or to assure software quality, patient safety, and data integrity? When regulated life sciences teams feel overwhelmed by documentation, the critical differences between Computer Software Assurance (CSA) and traditional Computer Software Validation (CSV) finally come to the surface. If you work in QA, compliance, or software deployment, this guide can help you determine which path best suits your environment.
Key takeaways:
-
Computer System Validation (CSV) remains a regulatory requirement and a fully compliant approach for demonstrating that software is fit for its intended use.
-
Computer Software Assurance (CSA) reflects the FDA’s current recommended approach for executing assurance activities for certain production and quality system software, not for all computerized systems universally.
-
CSA allows risk-based testing approaches, including unscripted testing for lower-risk functions where justified.
-
CSA does not replace CSV; it refines how validation activities are planned, executed, and documented.
-
How CSA principles are applied depends on system risk, update frequency, and an organization’s maturity in risk management and supplier oversight.
CSV vs CSA
Computer Software Validation (CSV)
Computer Software Validation refers to the regulatory requirement to establish and document that software specifications conform to user needs and intended uses through examination and objective evidence throughout the software lifecycle.
CSV has always allowed:
-
risk-based validation,
-
supplier involvement,
-
lifecycle management,
-
and proportional testing.
However, in practice, CSV has often been implemented through highly structured documentation, extensive scripted testing, and conservative interpretations, frequently driven by inspection risk aversion rather than documented failure modes or actual system risk.
Computer Software Assurance (CSA)
Computer Software Assurance is a risk-based approach recommended by the FDA for establishing and maintaining confidence that software used in production or quality systems is fit for its intended use.
CSA focuses on how validation activities are executed, emphasizing a least-burdensome approach where the level of assurance is proportional to the potential impact of software failure on product quality or patient safety. CSA applies specifically to computers and automated data processing systems used in medical device production and quality systems, and by extension informs similar thinking in pharmaceutical environments.
Regulatory context: how CSV and CSA relate
The relationship between CSV and CSA is defined by the FDA’s guidance Computer Software Assurance for Production and Quality System Software, which supersedes Section 6 of the older General Principles of Software Validation guidance.
Importantly:
-
CSV remains the regulatory obligation.
-
CSA clarifies FDA expectations for executing assurance activities for production and quality system software.
-
CSA does not eliminate validation requirements, lifecycle controls, or documentation expectations.
Comparison
The relationship between these two concepts is defined by the FDA, which issued the Computer Software Assurance for Production and Quality System Software guidance on September 24, 2025, to supersede Section 6 of the older General Principles of Software Validation guidance. [1,2]
|
Feature |
Computer Software Validation (CSV) |
Computer Software Assurance (CSA) |
|
Scope/focus |
General validation principles |
Focuses specifically on software assurance for production or the quality system for medical devices. |
|
Core philosophy |
Highly documented verification and testing throughout the software life cycle. |
A risk-based approach focused on the least burdensome effort. |
|
Risk determination |
The effort level is based on the safety risk (hazard) posed by the automated functions of the device. |
Assurance activities are assessed if they pose a high process risk. |
|
Testing/assurance rigor |
Detailed software testing, inspections, and static/dynamic analyses. |
Unscripted testing and external validation records. |
|
Current status |
The core guidance remains applicable to device software, but the section on validating automated process equipment and quality system software is superseded. |
Represents the FDA’s current recommendations for assuring computers and automated data processing systems used in production or quality systems. |
CSV: Limitations and challenges in modern digital environments
CSV was built for a different approach – one where software changed slowly, and updates happened once every few years. Technology evolved, but the CSV model stayed the same.
SaaS update cadence
Traditional CSV mandates that when any change is made to the software, the validation status of the software needs to be re-established. This requires a validation analysis to determine the extent and impact of the change on the entire system, followed by appropriate regression testing.
In a modern Software as a Service (SaaS) environment, updates are frequent. The requirement of a full re-validation and extensive scripted testing for every change becomes a burden.
Multi-system digital ecosystems
Modern environments often involve complex digital ecosystems where software features may be integrated and involve multiple systems. This includes cloud computing models like:
- Infrastructure as a Service (IaaS) – provides virtualized computing resources so companies don’t need to manage physical hardware;
- Platform as a Service (PaaS) – offers a ready-to-use environment for building and deploying applications without handling the underlying infrastructure;
- SaaS – delivers fully developed software applications over the cloud that users can access instantly without installation or maintenance.
CSV applies to systems composed of software components from many sources, such as:
- in-house developed software;
- off-the-shelf software;
- contract software.
The strict regulatory requirements for validation documentation mean the device manufacturer must assess the adequacy of the developer’s activities. This is important, especially for the Off-the-Shelf (OTS) software. Then they have to determine what additional efforts are needed to establish validation for their intended use.
This complexity results from the fact that necessary validation information or proprietary documentation (such as source code or vendor life-cycle documents) is frequently unavailable from commercial equipment vendors.
Complex, distributed validation burdens
CSV requires that validation processes and tasks remain the ultimate responsibility of the device manufacturer. They can be dispersed at different locations and conducted by different organizations; however, the responsibility falls on the manufacturer. For software components purchased OTS, the device manufacturer is responsible for ensuring the vendor’s methodologies are appropriate and sufficient.
The manufacturer may need to conduct sufficient black-box testing alone when vendor information is unavailable. The black-box testing is a software testing method where you evaluate what the system does without knowing anything about how it works internally. This placed an extremely heavy problem on the device manufacturer to solve. It is known that CSV decreases the validation efforts by 30%. [3]
Increased strictness: Why qualification scripts grew under CSV
The inclusion of Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) terminology in validation guidance promoted a rigid structure where validation tasks at the user site often involved extensive test cases with predefined expected results.
- IQ verifies that the system is installed correctly according to the manufacturer’s specifications.
- OQ confirms that the system functions as intended under defined operating conditions.
- PQ proves that the system consistently performs as intended in the real production environment.
This led manufacturers to often perform unnecessary and highly redundant validation, particularly for lower-risk functions. As a result, there were inflated OQ/PQ scripts and excessive focus on the number of tests rather than targeted risk, often failing to address the true system performance issues.
The accumulation of compliance debt
The complexity and documentation requirements of CSV often resulted in manufacturers either delaying validation or performing incomplete validation for software systems. The sources acknowledge that failure to document and archive test cases can significantly increase the level of effort and expense of revalidating the software.
This cumulative drag of delayed validation and incomplete documentation – the “compliance debt” – was a widespread issue stemming from the overly burdensome nature of CSV.
How CSA responds to these CSV limitations
CSA vs CSV is a natural answer to the strict policy for verification. CSA answers the limitations of CSV by shifting validation from a paperwork exercise to a business enabler. Teams reclaim time, budgets, and expertise that were previously locked inside endless documentation loops.
What makes this shift even more powerful is how naturally CSA fits the modern tech stack. Cloud platforms and SaaS applications no longer trigger panic or mountains of rework.
CSA lets organizations lean on trusted vendor evidence and concentrate their own effort only on the pieces that are customized or critical. This makes cloud adoption smoother and far more budget-friendly. Automation, intelligent analytics, bots, and AI tools become practical, low-friction additions to production and quality environments. CSA encourages the use of tools to automate testing and assurance activities, maximizing efficiency.
Importantly CSA does not reduce accountability; it redirects effort toward meaningful control and confidence.
CSV vs CSA in real use cases
|
Use Case |
CSV Approach (General Principles of Validation) |
CSA Approach (Risk-Based Assurance) |
|
SaaS QMS Monthly Updates |
Validation status must be re-established after any change to the software, even small ones. |
Low-risk updates rely on vendor evidence and targeted checks. Assurance efforts are scaled to address the risk. |
|
MES (Manufacturing Execution System) Module Changes |
A minor workflow adjustment requires extensive scripted testing across unrelated modules. |
Only the high-risk functions tied to product quality are tested in depth. Non-critical modules use unscripted testing or digital evidence for faster go-live. |
|
Laboratory Information Management System (LIMS) Vendor Patching |
A simple vendor-issued patch triggers broad revalidation. |
Teams classify the patch risk and perform focused assurance only if critical workflows (calculation steps, data integrity controls) could be affected. |
|
Integrations between systems |
Requires extensive planning and documentation. |
Focus on the intended use of the specific data transfer or control function. |
|
Custom scripts vs configurable systems |
Custom scripts necessitate rigorous verification activities like code inspection and traceability analysis. Commercial OTS requires the manufacturer to assess vendor development adequacy and supplement with user-site testing. |
Low-risk, configured functions may require assurance only via vendor risk assessment and installation checks. |
|
Analytics dashboards & CPV platforms |
Any software used to automate testing or part of the quality system must be validated for its intended use. |
If the function is for monitoring and review purposes that do not directly impact production or system performance, the manufacturer can rely on methods like exploratory testing. |
CSV vs CSA – risk profiles and when each makes sense

Computer System Validation remains the regulatory requirement in all cases. CSA does not introduce a separate compliance pathway, nor does it redefine when validation is required. What it changes is how assurance effort is allocated within that requirement.
In practice, CSA principles add the most value in environments where software behavior is not uniform across the system. Configurable platforms—particularly those updated frequently—often contain a mix of high- and low-risk functionality. Treating every feature as equally critical in such environments leads to disproportionate effort and limited insight into real failure modes.
CSA is particularly effective when:
-
systems are largely configurable rather than fully custom-built,
-
updates occur regularly, as with SaaS platforms,
-
individual functions vary significantly in their potential impact on product quality or patient safety,
-
and supplier quality systems are sufficiently mature to support reliance on vendor assurance activities.
In these contexts, CSA enables validation teams to focus depth where it matters most, while maintaining control over the system as a whole.
By contrast, more traditional validation execution may remain sufficient—and sometimes appropriate—for systems with direct patient-critical impact, highly customized control logic, or limited transparency into supplier development and testing practices. In such cases, the risk profile itself justifies more structured, tightly controlled assurance activities.
What FDA expects today
The FDA’s current posture toward software validation is not defined by new regulatory requirements, but by greater clarity around regulatory intent. Across recent guidance and inspection trends, the emphasis is consistently on whyassurance activities are performed, not merely how much evidence is produced.
Inspectors increasingly look for rational justification grounded in risk assessment, supported by objective evidence that is sufficient to demonstrate confidence in system performance. Excessive documentation that does not contribute to assurance is no longer viewed as inherently protective.
Both CSV and CSA must continue to satisfy the same core expectations: software must be fit for its intended use, quality system requirements must be met, and electronic records and signatures must comply with 21 CFR Part 11 where applicable.
Within that framework, the FDA has made clear that flexibility in testing methods is acceptable. Scripted testing, unscripted testing, or hybrid approaches may all be appropriate, provided the selected method is justified by the risk associated with the software function and the assurance objective being addressed.
Integrating CSA principles into existing validation frameworks
Adopting CSA does not require organizations to abandon their existing validation frameworks or start from a blank slate. Instead, it involves refining how decisions are made and documented within established quality systems.
In practice, this often means updating SOPs to explicitly reflect risk-based decision-making, clarifying how supplier evidence is evaluated and relied upon, and ensuring alignment between QA, IT, and business stakeholders on what constitutes sufficient assurance.
Equally important is training. Teams must understand not only what CSA allows, but why it reduces burden without reducing control. Without shared understanding and clear governance, increased flexibility can introduce inconsistency rather than improvement.
CSA therefore places greater emphasis on professional judgment. Strong QA oversight becomes more—not less—important, as validation decisions rely increasingly on documented rationale rather than predefined templates.
How CSA affects stakeholders
The shift toward CSA-aligned validation practices changes how different stakeholders engage with the validation lifecycle.
QA reviewers move away from primarily verifying the presence of predefined artifacts and toward evaluating the soundness of risk assessments and assurance rationale. Their role becomes less about counting documents and more about confirming that risks are understood and appropriately controlled.
IT system owners particularly those responsible for cloud-based and SaaS platforms gain greater flexibility in managing updates and configurations, while remaining accountable for maintaining validated states.
Regulatory teams benefit from closer alignment with the FDA’s least-burdensome philosophy, as CSA provides a clearer framework for explaining and defending validation decisions during inspections.
Engineering teams redirect effort from highly formal documentation toward targeted testing and system understanding, improving both efficiency and software quality.
Vendors, in turn, take on a more visible role in the validation ecosystem. Suppliers with transparent development practices, robust quality systems, and well-documented assurance activities become strategic partners rather than opaque dependencies.
Choosing the right path: A decision framework
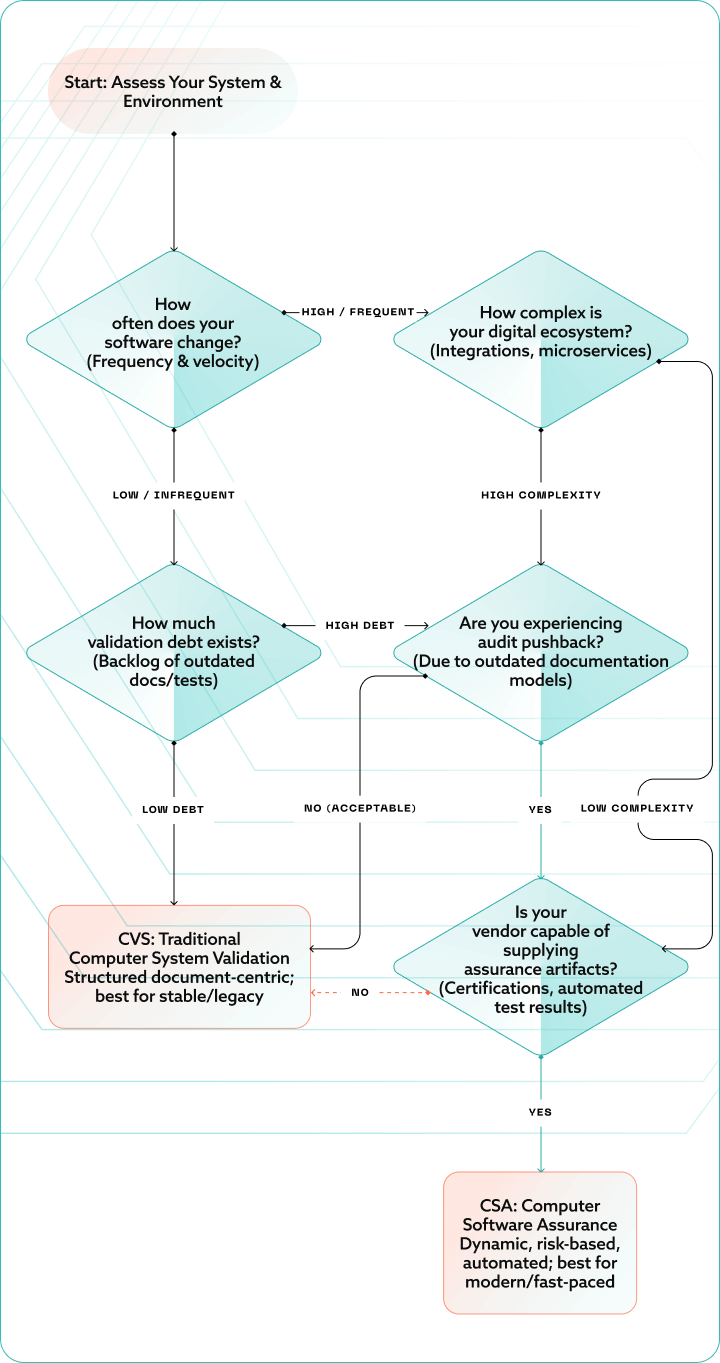
Frequently Asked Questions (FAQ)
Is CSV still acceptable to the FDA?
Yes, CSV remains fully acceptable and compliant for validating computerized systems. The FDA recognizes its continued relevance alongside newer approaches.
Does CSA replace CSV entirely?
No, CSA does not completely replace CSV; it is an alternative approach designed for modern and higher-risk systems. Organizations may use either method depending on their system needs.
Is CSA faster and cheaper than CSV?
CSA can be faster and more cost-effective. Efficiency gains depend on system complexity and update frequency therefore CSV is still a viable option.
Will auditors require CSA over CSV?
Not necessarily; auditors accept both approaches as long as they are justified and documented appropriately. CSA may be preferred for modern systems, but CSV is still fully compliant.
Do all systems qualify for CSA?
No, CSA is best suited for systems with frequent updates and sufficient process maturity. Low-risk or legacy systems may continue using traditional CSV.
Conclusion
Computer System Validation is not disappearing. It remains the foundation of computerized system compliance and a critical mechanism for protecting product quality and patient safety.
Computer Software Assurance does not replace CSV. It clarifies how validation should be executed in modern software environments, where systems evolve continuously and risk is not evenly distributed.
Together, CSV and CSA represent two perspectives on the same objective: maintaining confidence that software performs as intended throughout its lifecycle. Organizations that understand this relationship are better positioned to modernize responsibly without increasing regulatory risk.
If you need support applying CSA principles within your existing validation framework, BGO Software can help translate regulatory intent into practical, inspection-ready execution.
References:
[1] U.S. Food and Drug Administration, Center for Devices and Radiological Health, & Center for Biologics Evaluation and Research. (2025, September 24). Computer Software Assurance for Production and Quality System Software: Guidance for industry and Food and Drug Administration staff. U.S. Department of Health and Human Services.
[2] U.S. Department of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, & Center for Biologics Evaluation and Research. (2002, January 11). General principles of software validation; Final guidance for industry and FDA staff [Guidance Document].
[3] Thirumalai, P. P. (2023). Computer software assurance – An interpretation and future [Unpublished manuscript]. Center of Excellence in Regulatory Science and Innovation, Johns Hopkins University. https://publichealth.jhu.edu/sites/default/files/2023-07/pavithra-thirumalai.pdf

Healthcare business analyst with expertise in marketing and business development, and holds an MPharm degree. He specialises in creating and executing communication strategies that make digital health solutions and pharmaceutical technologies clear, accessible, and resonation for their audiences.
link to the author’s linkedin profile



