
Most pharmaceutical manufacturers spend between 3 and 6 months preparing a single Annual Product Quality Review. For a site producing dozens of products, that’s thousands of person-hours moved into spreadsheets every year. The work itself is mostly mechanical: pull data from LIMS, MES, ERP, and QMS, reconcile timestamps, reformat into the corporate template, and write up trends. The actual quality assessment, the part regulators care about, gets squeezed into whatever time is left.
AI is being pitched as the fix for this. The pitch is partially right. The interesting part is what has to be true before the fix works, and how it fits into GMP-validated systems without breaking compliance.
What is APQR? The Annual Product Quality Review (APQR), also called Product Quality Review (PQR) under EU GMP or Annual Product Review (APR) under FDA 21 CFR 211.180(e), is a yearly assessment of every manufactured product. It confirms manufacturing process consistency, evaluates deviations and CAPAs, reviews stability data, and identifies whether changes are needed to specifications, processes, or controls.
Key Takeaways
- APQR is a data problem first, an analysis problem second. Around 65% of total APQR effort goes into collecting and reconciling data across LIMS, MES, ERP, QMS, and historians. Automation only pays back if that foundation is fixed first.
- Contextualisation is the step most teams skip. ISA-88 and ISA-95 give you the vocabulary, but internal definitions (batch start, deviation linkage, site mapping) must be aligned before any AI tool produces trustworthy output.
- Agentic AI is compliant when validated as a chain. Under EMA Draft Annex 22, validation covers the data sources, contextualisation rules, prompt templates, and human review step. You are not validating the LLM, you are validating the workflow.
- Human-in-the-loop is non-negotiable. The agent prepares the draft. The accountable reviewer signs off. The model mirrors the sponsor-CDMO accountability split regulators already accept.
- The same data foundation serves more than APQR. Once built, it supports CPV, environmental monitoring, deviation prediction, and yield optimisation. The annual report becomes a snapshot from a system already monitoring everything continuously.
The Real Cost of a Traditional APQR
Industry analysis puts manual APQR preparation at 3 to 6 months of QA effort per product. The FDA issued 149 warning letters across compliance programs and recalls covering 290 drug products in a single year, with many findings traceable to weak APQR documentation or inconsistent trending.
The hours add up because the data lives in too many places. Here is where a single batch leaves traces, and what the APQR author has to reconcile across them.
|
Data source |
What it holds |
Why it complicates APQR |
|
QMS |
Deviations, complaints, CAPAs, change controls |
Batch linkage often weak or manual |
|
LIMS |
Release testing, stability, COA data |
Timestamps rarely align with batch phases |
|
MES / Electronic Batch Record |
Process steps, parameters, operator actions |
Paper-based sections still common |
|
ERP |
Materials, suppliers, batch genealogy |
Different batch naming conventions |
|
Historian |
Real-time process data, environmental conditions |
Tied to equipment, not always to batch |
|
Stability systems |
Long-term and accelerated data |
Separate IT system, separate access controls |
Stitching this together by hand is where the months go. Around two-thirds of total APQR effort is data acquisition and reconciliation, not analysis. The actual quality story, the part the regulator pays attention to, gets the leftover time. Our optimizing APQR in pharma guide covers the manual workflow breakdown in more detail.
Why APQR Became a Bureaucratic Exercise
The original intent was the opposite. The FDA codified the Annual Product Review in 21 CFR 211.180(e), effective March 1979. EU GMP added the Product Quality Review in Chapter 1 of EudraLex Volume 4 in 2006. ICH Q7 extended the same expectation to active pharmaceutical ingredients, and ICH Q10 embedded it into the Pharmaceutical Quality System.
Across all four frameworks, the intent is the same. The annual review should drive continuous knowledge extraction from a year of manufacturing data. According to peer-reviewed work on pharmaceutical Quality by Design (Yu et al., AAPS Journal), the goal is process understanding tight enough to predict and prevent failures, not just document them.
The reality in most companies is different. APQR has drifted into a yearly compliance scramble. Issues get logged but rarely operationalised. Improvement opportunities surface but get parked because the team is already three months behind on next year’s review. Auditors notice. Common 483 observations include:
- Lack of traceability between deviations and CAPAs
- Inadequate trending of out-of-specification results
- Fragmented change control documentation
- Missing justification for batches that fell outside normal limits
- No effectiveness check on previous CAPAs
The 80/20 Data Problem
The pattern that shows up in every AI project in pharma also shows up in APQR work. Roughly 80% of effort goes into data preparation. The analytical work is the last 20%. The implication is uncomfortable but worth stating clearly: if your data foundation is not in shape, no AI tool will save you. Agentic AI included.
Here is how the time actually splits inside a typical APQR cycle.
|
Activity |
Share of total effort |
What goes wrong |
|
Data collection across source systems |
60-65% |
Manual exports, format mismatches, missing context |
|
Formatting and writing |
15-20% |
Copy-paste errors, template version drift |
|
Statistical analysis and trending |
10-15% |
Time-boxed, run on incomplete data |
|
Review and approval |
5-10% |
Late-cycle, under deadline pressure |
The same logic applies to healthcare data integration more broadly. Whether the output is an APQR, a continued process verification dashboard, or a deviation prediction model, the data foundation is the precondition for everything else.
Data Contextualisation: The Step Nobody Talks About
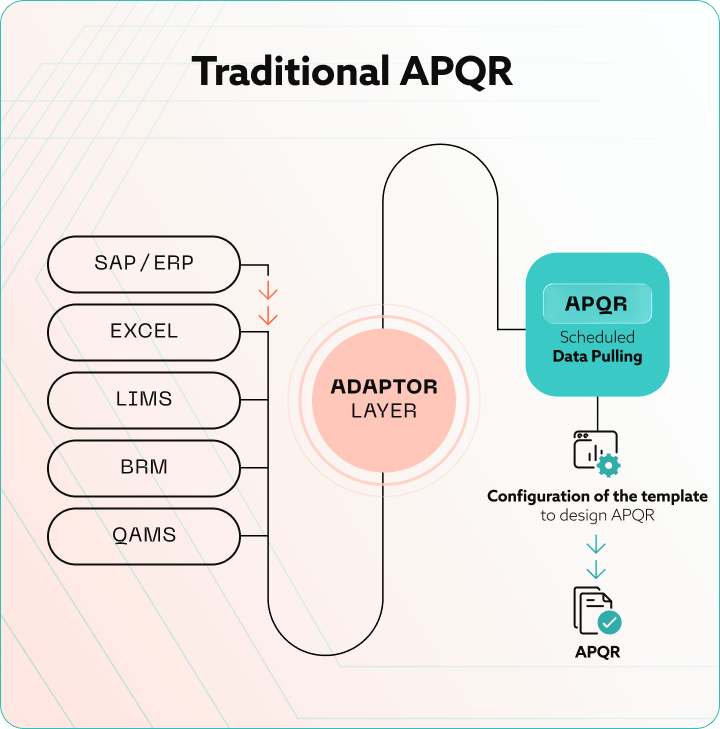
The single most underrated step in any APQR automation effort is data contextualisation. The industry has standards for this. ISA-88 defines the batch model. ISA-95 defines the hierarchy of enterprise, site, area, work centre, and work unit. Together they give you a vocabulary for tagging every data point with the batch, phase, equipment, and operation it belongs to.
Standards alone do not solve the problem. The harder issue is internal consistency.
Take a concrete example. A site runs a filling line followed by a packaging line. The batch crosses both. When does the batch start? Some teams define the start as the first vial entering the filling line. Others define it as the first packaged unit exiting the packaging line. Both definitions are defensible. What is not defensible is having both in use inside the same company without anyone reconciling them. Run an AI model across that dataset and it will produce confident, well-formatted, plausible-looking nonsense.
The same problem shows up with deviations that do not link cleanly to a batch phase, customer complaints in a QMS that does not require a site field, and environmental monitoring data captured at the room level but never tied to the specific batch operation that was running. These are not exotic edge cases. They are the daily reality of pharmaceutical data. A Pharma Data Management Solution that breaks the silos and applies consistent contextualisation rules across LIMS, MES, ERP, and QMS data is the precondition for everything that follows.
How AI Fits Into APQR Without Breaking GMP
The most common objection to agentic AI in a regulated environment is that large language models are non-deterministic, and regulators are uncomfortable with that. Fair point. It is also a reason to think carefully about where in the workflow the AI sits, not a reason to dismiss it.
The EMA Draft Annex 22 on Artificial Intelligence (July 2025) lays out the expectations: intended use must be defined, validation must follow lifecycle principles, explainability is required, and human-in-the-loop oversight is non-negotiable for GxP-critical decisions. The follow-up peer-reviewed interpretation of Annex 22 (Stassen et al., PDA Journal, 2026) maps these expectations to operational practice.
A useful analogy comes from contract manufacturing. A sponsor outsources production to a CDMO. The CDMO has the data, runs the process, and prepares the draft APQR. The sponsor reviews, challenges, and approves. The sponsor remains the accountable party. Nobody argues this arrangement is non-compliant.
Agentic AI plays the same role as the CDMO in that analogy. The table below shows the split.
|
Step in APQR workflow |
AI does |
Human reviewer does |
|
Define prompt instructions from SOP |
— |
Writes, validates, version-controls |
|
Pull data from validated sources |
Executes against approved connectors |
Approves source list |
|
Apply contextualisation rules |
Tags batch, phase, equipment |
Confirms rules match SOP |
|
Populate template sections |
Fills tables, charts, trend summaries |
Reviews against source data |
|
Flag anomalies and trends |
Surfaces statistical outliers |
Decides if action is required |
|
Final sign-off |
— |
Accountable QP signature |
This reframes the validation question. You are not validating an LLM. You are validating a chain that includes data sources, integration logic, contextualisation rules, prompt templates, and a documented human review step. Each link is something the industry already knows how to validate. Our GAMP 5 second edition coverage explains how the risk-based validation framework adapts to AI-enabled systems. The empirical evidence is already building, with peer-reviewed work on LLM-assisted deviation investigation (Salami et al., 2024) and scalable deep learning for real-time multivariate batch monitoring (Sammaknejad et al., 2025) inside major pharma manufacturing environments.
What AI Actually Delivers for an APQR

When the data foundation is in place, AI collapses the mechanical layer of APQR work into minutes. The agent pulls validated data from each source system, contextualised to the right batch and phase, and populates the template directly. Multivariate analysis flags out-of-trend results, correlations between critical process parameters and critical quality attributes, and emerging drift. Deviations link to batch phase, change controls, and CAPA effectiveness checks. Every data point pulled and every flag raised is logged for 21 CFR Part 11 and Annex 11 audit trails.
The same infrastructure supports continued process verification, environmental monitoring, batch comparison, and yield optimisation. The annual report becomes a snapshot pulled from a system already monitoring everything in real time. Some practitioners call this a Continuous Product Quality Review (CPQR). The label matters less than the change in workflow.
The shift from manual to agentic-assisted APQR looks like this in practice.
|
Dimension |
Manual APQR |
AI APQR |
|
Time per product |
3-6 months |
Hours to days |
|
Effort split |
65% collection, 35% analysis |
15% review, 85% analysis |
|
Data freshness |
Annual snapshot |
Continuous |
|
Audit trail |
Reconstructed at year-end |
Generated in real time |
|
Anomaly detection |
Single-variable, manual |
Multivariate, automated |
|
Reusability of infrastructure |
Single output |
CPV, EM, deviations, yield |
A drug manufacturing intelligence implementation for a top 5 pharmaceutical company in Switzerland shows what this looks like in practice: real-time process and product monitoring, continued process verification, and standardised trending across manufacturing sites, all on a GMP-validated platform.
What to Do Next Week
Three things are worth doing regardless of whether you ever deploy an agentic AI tool.
Audit your APQR cycle. Measure how many hours per product go into data collection, formatting, analysis, and review. The ratio tells you exactly where automation will pay back and where it will not.
Pick one contextualisation conflict in your data and fix it. Batch start definitions across lines. Deviation linkage to batch phase. Customer complaint to site mapping. Whatever the worst one is, write down the canonical definition and start enforcing it in new data.
Translate one APQR section of your SOP into prompt-style instructions. Not because you are about to deploy an agent next month, but because the exercise will surface the implicit decisions your reviewers are making that nobody has written down. Those decisions are exactly what an agent will need, and what the audit trail of any future automation will require.
The industry conversation about agentic AI has moved faster than most quality teams have had time to absorb. The underlying mechanics have not. APQR is a data problem with an analysis layer on top. Solve the data problem and the analysis layer takes care of itself, with or without an agent. Skip the data problem and the agent will produce three months of confident output that nobody can trust.
If you want to map this to your own pharmaceutical quality workflow, our GMP-validated systems team and regulatory and compliance specialists can help you scope the data foundation and validation chain.
Sources & Further Reading
Regulatory & Guidance Documents
-
S. Food and Drug Administration. 21 CFR 211.180 — Current Good Manufacturing Practice for Finished Pharmaceuticals. Code of Federal Regulations, Title 21.
-
European Commission. EudraLex Volume 4, Chapter 1: Pharmaceutical Quality System. The Rules Governing Medicinal Products in the European Union, 2013.
-
European Commission. Draft EU GMP Annex 22: Artificial Intelligence. Consultation Document, July 2025.
-
International Council for Harmonisation. ICH Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. 2000.
-
International Council for Harmonisation. ICH Q10: Pharmaceutical Quality System. 2008.
-
International Council for Harmonisation. ICH Q8(R2): Pharmaceutical Development. 2009.
-
S. Food and Drug Administration. Artificial Intelligence in Drug Manufacturing — Discussion Paper. CDER, March 2023.
-
Pharmaceutical Inspection Co-operation Scheme. PIC/S PI 009-17 Part I: Guide to Good Manufacturing Practice for Medicinal Products. 2023.
Peer-Reviewed Research
-
Yu LX, Amidon G, Khan MA, et al. Understanding pharmaceutical quality by design. The AAPS Journal, 16(4):771-83, 2014.
-
Ondracka A, Gasset A, García-Ortega X, et al. CPV of the Future: AI-Powered Continued Process Verification for Bioreactor Processes. PDA Journal of Pharmaceutical Science and Technology, 77(3):146-165, 2022.
-
Stassen M, Leitão CS, Manzano T, Valero F, et al. Recommendations for Artificial Intelligence Application in Continued Process Verification. PDA Journal of Pharmaceutical Science and Technology, 79(1):68-87, 2025.
-
Stassen M, Schmucki M, Valero F, et al. Bridging Guidance and Regulation: Interpreting the Draft Annex 22 on Artificial Intelligence in GMP Manufacturing. PDA Journal of Pharmaceutical Science and Technology, 80(2):248-254, 2026.
-
Salami H, Smith-Goettler B, Yadav V. How can language models assist with pharmaceuticals manufacturing deviations and investigations? International Journal of Pharmaceutics, 668:125100, 2024.
-
Sammaknejad N, Lee J, Austria JM, et al. A Scalable Deep Learning Approach for Real-Time Multivariate Monitoring of Biopharmaceutical Processes. Biotechnology and Bioengineering, 122(9):2333-2352, 2025.
-
Huanbutta K, Burapapadh K, Kraisit P, et al. Artificial intelligence-driven pharmaceutical industry: A paradigm shift in drug discovery, formulation development, manufacturing, quality control, and post-market surveillance. European Journal of Pharmaceutical Sciences, 195:106938, 2024.
Industry Standards
-
International Society of Automation. ANSI/ISA-88 (IEC 61512): Batch Control.
-
International Society of Automation. ANSI/ISA-95 (IEC 62264): Enterprise-Control System Integration.
-
Baseline Guide Volume 8: Pharma 4.0™. International Society for Pharmaceutical Engineering, 2023.
-
GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems, Second Edition.
Pharmaceutical Inspection Co-operation Scheme. PIC/S PI 041: Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments

Healthcare business analyst with expertise in marketing and business development, and holds an MPharm degree. He specialises in creating and executing communication strategies that make digital health solutions and pharmaceutical technologies clear, accessible, and resonation for their audiences.
link to the author’s linkedin profile