


Pharmaceutical manufacturing has operated on batch principles for well over a century. Ingredients move through discrete, sequential stages each stopping before the next begins. Materials wait in holding tanks, get shipped between facilities, and spend weeks accumulating toward a finished product that could, in many cases, be produced in days.
Continuous pharmaceutical manufacturing (CPM) closes that gap. It is an “always-on” process that runs raw materials through a single, uninterrupted line all the way to finished product eliminating the hold times, inter-stage delays, and batch-related inefficiencies that define traditional production. The result is higher output, lower costs, improved quality through real-time monitoring (PAT tools), and a faster path from process to market.
That gap between what is possible and what is practiced is what makes CPM worth understanding not as a trend, but as a production decision with concrete technical, regulatory, and economic dimensions.
Key Takeaways:
- Continuous pharmaceutical manufacturing (CPM) is an always-on production method that runs raw materials through to finished product on a single uninterrupted line, eliminating the hold times, process stoppages, and inter-facility transfers that routinely stretch batch production cycles into months increasing output, reducing costs, and enabling real-time quality monitoring through PAT tools that end-of-batch testing cannot match.
- The FDA’s own self-audit found that continuous manufacturing applications had shorter approval times and earlier market entry than comparable batch submissions, translating to an estimated $171–537 million in early revenue benefit per product, directly contradicting the widespread assumption that regulatory risk is higher for continuous processes.
- Successful continuous manufacturing implementation depends on GxP-validated digital infrastructure validated MES, full audit trails meeting ALCOA+ standards, and unified process data architecture because without it, the volume of real-time data a continuous line generates cannot be used for regulatory-grade decision-making or real-time release testing.
- The business case for switching from batch to continuous is product-specific. Existing products on stable batch processes with low volume or short remaining patent life rarely justify the conversion cost, while new products and high-volume oral solid dosage forms present the strongest return on investment.
- AI-assisted process control is moving from pilot projects into routine use, with anomaly detection systems identifying deviations in seconds rather than minutes which is a difference that directly determines how much material is lost or diverted when a critical process parameter moves outside its validated range.
Pharmaceutical manufacturing has operated on batch principles for well over a century. Ingredients move through discrete, sequential stages. Each stage stops before the next begins. Materials wait in holding tanks, get shipped between facilities, and spend weeks accumulating toward a finished product that could, in many cases, be produced in days.
That gap between what is possible and what is practiced is what makes continuous pharmaceutical manufacturing (CPM) worth understanding, not as a trend, but as a production decision with concrete technical, regulatory, and economic dimensions.
What continuous pharmaceutical manufacturing actually is?
CPM is a production method in which input materials enter and output materials exit simultaneously, with all unit operations connected into a single, uninterrupted flow. Quality testing happens inline through Process Analytical Technology (PAT) rather than at the end of each stage.
This differs fundamentally from batch processing, where each step must be completed before the next begins. In batch manufacturing, a drug product traveling through six or seven stages may spend the majority of its production time sitting idle between steps. Those hold times create compounding risk: material degradation, supply chain dependency, and quality variability that only surfaces at end-of-batch testing.
The MIT-Novartis collaboration demonstrated what this difference looks like in practice. An end-to-end continuous process for aliskiren hemifumarate tablets produced finished product from API starting materials in approximately two days. The equivalent batch process, accounting for individual step completion, material release testing, and inter-facility transport, can take up to 12 months.
Batch vs continuous pharmaceutical manufacturing: the structural difference
|
Factor |
Batch manufacturing |
Continuous manufacturing |
|
Lot size |
Defined by equipment volume |
Defined by run time or mass flow rate |
|
Scale-up method |
Larger equipment, revalidation |
Run longer at same parameters |
|
Quality control |
End-of-batch testing |
Real-time inline monitoring (PAT) |
|
Hold times |
Multiple between stages |
Eliminated or minimized |
|
Response to demand change |
Equipment-dependent |
Adjust run duration |
|
Changeover complexity |
Lower |
Higher; can take days to a week |
|
Upfront investment |
Lower |
Substantially higher |
Lot size flexibility deserves specific attention. In batch manufacturing, increasing production volume for an approved product requires larger equipment, scale-up validation studies, and a regulatory submission for the change. In continuous manufacturing, the same process runs at the same parameters for longer. Post-approval volume changes are substantially simpler because the process parameters defining quality remain constant.
How continuous flow chemistry changes API synthesis
For small molecules, continuous flow chemistry in pharmaceutical manufacturing has demonstrated advantages that batch reactors cannot replicate:
- PFRs and PBRs operate across −80°C to 300°C, versus −20°C to 120°C for standard batch vessels
- Smaller reactor volumes reduce exposure to hazardous reagents by orders of magnitude
- Reactive gases such as hydrogen, carbon monoxide, and ammonia can be handled under higher pressure than batch equipment allows
- Unstable intermediates can be generated and immediately consumed without accumulation
Eli Lilly’s continuous cGMP process for prexasertib monolactate monohydrate — an oncology compound ran eight unit operations in series across four synthetic route steps at 3 kg per day. The hydrazine reaction forming a pyrazole intermediate used significantly less hydrazine than a comparable batch process and held only 1.4 liters of the reagent in the PFR at any time. Three coiled tube reactors made from PFA were inexpensive enough to dispose of between campaigns, removing cross-contamination risk without cleaning validation.
Novartis, which began investing in continuous API manufacturing in 2007, replaced two 10,000-liter batch reactors with a 16-liter PFR for one metalation reaction and improved yield by 50% through continuous countercurrent extraction with reagent recycle. Their multipurpose continuous plant now operates at up to 15 tons per year of API under cGMP.
Regulatory approval: slower for continuous manufacturing, or faster?
One of the most persistent misperceptions about continuous manufacturing of pharmaceuticals is that regulatory approval is slower or harder to obtain. The FDA’s own self-audit found the opposite.
Applications using continuous manufacturing had shorter times to approval and earlier market entry than comparable batch applications, translating to an estimated $171–537 million in early revenue benefit per product, with no substantial regulatory barriers identified related to manufacturing process changes or pre-approval inspections.
The approval timeline tells a clear story:
- 2015 — Vertex Pharmaceuticals’ Orkambi (lumacaftor/ivacaftor), first FDA approval for a continuously manufactured drug product
- 2016 — Janssen’s Prezista (darunavir), first approval for a batch-to-continuous conversion on an existing product
- 2016–2018 — Eli Lilly and Pfizer received approvals for additional continuously manufactured products
ICH Q13, implemented in Europe in July 2023, provides a comprehensive regulatory framework for continuous manufacturing of pharmaceuticals, addressing batch definition, control strategy, material traceability, and diversion mechanisms for non-conforming material. Both the FDA and EMA now publish detailed guidance for continuous setups, including real-time release testing and model-based control strategies.
PAT and Quality by Design: the operational core
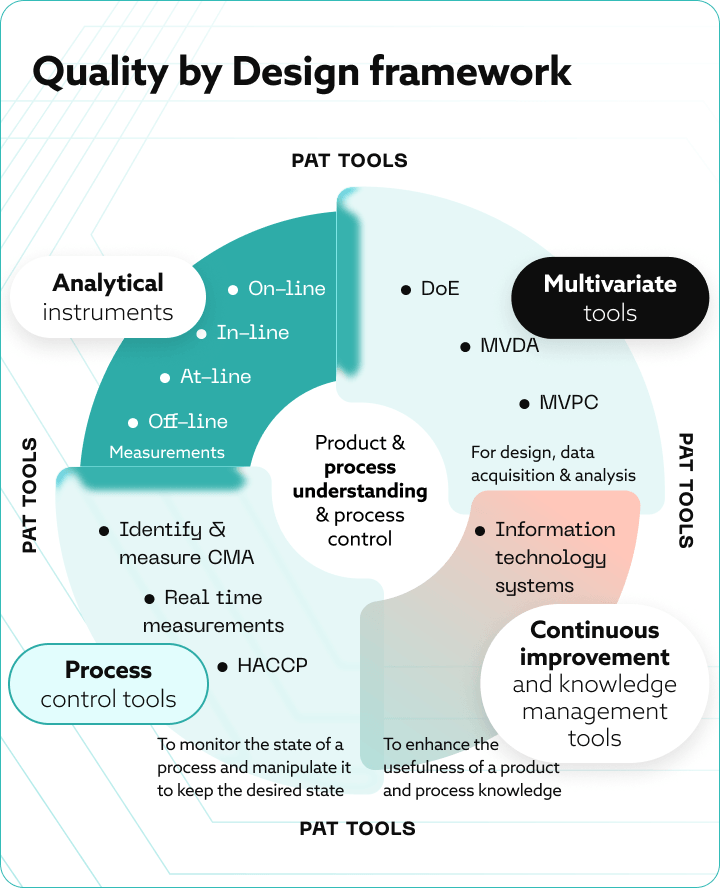
CPM is inseparable from PAT. The two developed in parallel, and neither reaches full function without the other.
The relationship sits within a broader Quality by Design (QbD) framework. One that places product and process understanding at the center, with four categories of tools surrounding it: analytical instruments for on-line, in-line, at-line, and off-line measurement, multivariate tools including Design of Experiments (DoE) and MVDA for data acquisition and analysis.
Also process control tools such as HACCP for monitoring and maintaining desired process states and continuous improvement and knowledge management systems that carry process learning forward across the product lifecycle.
PAT tools operate across all four quadrants, which is what the framework diagram below illustrates.
In practice, this means PAT tools near-infrared spectroscopy, Raman spectroscopy, inline HPLC, mass flow measurement monitor Critical Quality Attributes (CQAs) and Critical Process Parameters (CPPs) throughout production rather than after it. QbD defines the design space during development: the CPP ranges within which product quality is reliably maintained. PAT verifies that production stays within that space in real time, and the process control layer acts on what PAT reports.
The Orkambi approval shows what this looks like when it is properly implemented. In-process controls used gravimetry at pre- and post-granulation blending stages, NIR spectroscopy for granule moisture and particle size, and Raman spectroscopy for drug substance form and film coat thickness. That data enabled a real-time release testing dissolution model, removing the need to wait for offline laboratory-based results before releasing the product.
The outcome is that GMP requirements are met through continuous data rather than retrospective batch testing which reduces time to product release and produces a more complete record of process behavior than periodic end-of-batch sampling can generate.
Challenges in continuous manufacturing of pharmaceuticals
The benefits above are real, but they do not arrive without friction. Three operational challenges consistently slow adoption.
Changeovers are the most immediate. Continuous manufacturing lines have thousands of interconnected components
reactors, sensors, flow paths, control systems that must be disassembled, cleaned, and verified when switching products. The target for the industry is changeover in under a day. Current lines routinely take a week or more. For facilities running multiple products on a single line, that time comes directly out of productive capacity.
Operator training is a related constraint. Continuous manufacturing equipment is more complex than batch equipment, and the consequences of errors propagate through an integrated system rather than staying contained within a single stage. Everyone involved in operating the line needs substantive exposure to how the system behaves under normal conditions and under deviation — not just procedural familiarity.
Economics is where many projects stall. Converting an existing product from a validated batch process to continuous manufacturing requires capital expenditure on new equipment, process development work, regulatory submission preparation, and — in some cases — bioequivalence studies. For a product already generating stable revenue on a batch process, that investment rarely justifies itself unless volume is high, demand is variable, or supply chain resilience is a strategic priority.
None of these are arguments against continuous manufacturing. They are constraints that affect which products and facilities make sense to convert first.
Getting continuous manufacturing to work: what the equipment alone cannot do
Switching to continuous manufacturing depends less on the equipment itself and more on the systems wrapped around it. Four capabilities consistently separate facilities that succeed from those that stall.
PAT integration gives operators visibility during production instead of after the fact. Tools measuring flow, temperature, pH, and particle size in real time allow deviation correction before a batch is lost rather than after it is tested.
Digital twins allow teams to test new process setups, predict CQA responses to parameter changes, and stress-test control strategies virtually before touching live equipment. They shorten design validation cycles and reduce risk when scaling or adjusting a qualified process.
Manufacturing Execution Systems (MES) that can be configured without extended IT development cycles matter because continuous manufacturing generates coordination requirements, material movement, electronic batch records, procedural enforcement across shifts that rigid, legacy MES platforms handle poorly. Facilities that bring Head of IT and QA/QC managers and Plant Heads, into the implementation from the start avoid untangling compliance and integration issues later.
Edge analytics and industrial IoT bring decision-making closer to the equipment. Sensors and edge devices collect data across the line; the relevant information reaches operators in time to act on it. The same infrastructure supports predictive maintenance and material traceability both of which matter when every hour of uptime affects output.
Where to begin: a grounded roadmap for continuous
Facilities that implement continuous manufacturing successfully tend to follow a consistent sequence rather than attempting full-facility conversion at once.
Step 1:
A genuine feasibility assessment, not an optimistic one. Pick a product or unit operation that runs at volume, has well-understood process parameters, and is mechanically suited to continuous flow — blending, granulation, and coating are common starting points. Run a pilot with real materials and proper instrumentation. Use it to validate process control logic, integrate PAT tools, and build the digital twin before committing to the full line.
Step 2:
Regulatory alignment, ideally started during the pilot rather than after it. Many implementations stall here not for technical reasons but because teams wait until they have data before engaging the FDA or EMA. Both agencies have indicated openness to early dialogue on continuous manufacturing submissions. PAT data from pilot runs is the most useful material for demonstrating process stability during those conversations.
Step 3:
The third step is scaled MES integration. Once a continuous line is validated, expansion is primarily a systems coordination challenge connecting material movement, digital SOPs, electronic batch records, and real-time process decisions across an increasingly complex line. Hybrid approaches, running continuous granulation alongside batch compression for example, are common intermediate configurations that allow modernization without reworking an entire facility simultaneously.
AI in continuous manufacturing of pharmaceuticals

A continuous manufacturing line generates more process data per hour than a batch facility generates in a week. AI-assisted process control is moving from pilot projects toward routine use:
- Systems trained on historical process data detect early deviation signatures — temperature drift, flow rate instability, NIR spectral shifts — before they propagate to out-of-specification material
- Predictive models identify CPP combinations that correlate with CQA failures, enabling targeted intervention rather than reactive correction
- Digital twins running in parallel with live production allow real-time comparison between predicted and observed process behavior
For continuous drug product processes, where mean residence times are measured in minutes, the speed of anomaly detection directly affects yield. An out-of-specification event that takes 30 minutes to detect in a continuous tablet line may affect 15 minutes of production. An event detected in 90 seconds affects a few thousand tablets. That difference has measurable material yield and regulatory consequence.
Modular facility design is developing alongside these capabilities. Plants built from skidded, reconfigurable units can be deployed faster for new products, replicated across geographies without extensive process transfer work, and reconfigured as portfolio needs change. When combined with digital twins, edge analytics, and well-integrated MES platforms, modular facilities reduce the capital commitment required to enter continuous manufacturing while preserving operational flexibility.
The integrated GxP software infrastructure this requires
Continuous pharmaceutical manufacturing does not function as an isolated technical upgrade. It requires validated software infrastructure, AI-driven analytics, and regulatory-grade lifecycle management functioning together within a GxP-compliant environment.
Digital systems operating in GxP environments must meet non-negotiable requirements: validated frameworks meeting CSV and CSA standards, full audit trails satisfying ALCOA+ data integrity requirements, role-based access and controlled workflows, and complete traceability across the validation lifecycle. Platforms implementing GMP-compliant multivariate data analytics (MVDA) have demonstrated 50% reductions in manual validation effort and significant reductions in protocol review and approval cycle times — but only when the separation between validated GxP modules and non-validated exploratory modules is properly designed.
Lot genealogy is more complex in continuous manufacturing than in batch. In batch processing, it means verifying that all charged components are mixed uniformly in the finished product. In continuous processing, feed streams are periodically recharged, potentially from different material lots, and tracking those components through the entire process train requires process flowsheet modeling and explicit decisions about how batches are defined by processing time or by volume of material produced.
When validated digital systems and AI-driven analytics work together within that framework, the outcomes are concrete: reduced validation cycle times, faster deviation detection, improved yield prediction through multivariate process modeling, and a foundation for real-time release testing that reduces the lag between production and product release.
The manufacturers who benefit most from continuous pharmaceutical manufacturing are those building not just continuous lines, but the integrated, GxP-compliant data ecosystems that allow those lines to be operated, monitored, and continuously improved within regulatory requirements. The technology is proven. The regulatory framework exists. What determines success now is organizational readiness and infrastructure design.
Still not sure where to start? Let’s discuss how we can support you.
Frequently Asked Questions (FAQ)
How long does FDA approval take for a continuously manufactured drug compared to batch?
The FDA’s own self-audit found continuous manufacturing applications reached approval and market entry faster than comparable batch submissions, with the advantage estimated at $171–537 million in early revenue per product. No substantial regulatory barriers specific to continuous manufacturing were identified. ICH Q13, implemented in Europe in July 2023, provides a unified framework covering batch definition, control strategy, and material traceability for continuous manufacturing submissions globally.
What is the difference between PAT monitoring and end-of-batch testing in pharmaceutical manufacturing?
End-of-batch testing evaluates a finished batch after production completes and releases it only if it meets specification — meaning any quality problem is discovered after the fact. PAT monitors Critical Quality Attributes and Critical Process Parameters in real time during production using tools such as near-infrared spectroscopy, Raman spectroscopy, and inline HPLC. Data feeds directly into process control systems, allowing corrections before deviations affect product quality. In the Orkambi cystic fibrosis approval, this approach enabled a real-time release testing dissolution model that replaced offline laboratory testing entirely.
What validated digital infrastructure does a continuous pharmaceutical manufacturing facility require?
A GxP-compliant continuous manufacturing environment requires validated frameworks meeting CSV and CSA standards, audit trails satisfying ALCOA+ data integrity requirements, role-based access controls, GMP-compliant multivariate data analytics, and an MES capable of handling electronic batch records and real-time procedural enforcement. Lot genealogy tracking is also non-negotiable — feed streams in continuous manufacturing are periodically recharged from different material lots, and tracking those components through the process train requires process flowsheet modeling that batch facilities do not need.
Does continuous pharmaceutical manufacturing work for biologics or primarily small molecules?
The strongest commercial track record is in small molecule oral solid dosage forms, where multiple FDA-approved continuously manufactured products have been on the market since 2015. Biologics manufacturing currently relies mostly on hybrid configurations continuous perfusion bioreactors upstream paired with batch purification downstream. Fully integrated end-to-end continuous bioprocessing has been demonstrated at development scale, but downstream chromatography synchronization complexity and the training requirements for integrated system operation have slowed commercial adoption.
Why do continuous pharmaceutical manufacturing implementations fail?
Three problems account for most stalled projects. Changeover complexity is the most immediate — disassembling, cleaning, and reassembling thousands of line components between products routinely takes a week or more, directly cutting into productive capacity. The second is treating digital infrastructure as secondary to equipment: facilities that delay PAT integration, MES configuration, and validated data architecture consistently encounter compliance gaps that block regulatory approval. The third is a poorly constructed business case for low-volume products or those late in their patent lifecycle, the capital, process development, and regulatory costs of conversion rarely produce a positive return.
Sources:
- Prexasertib
- Continuous Manufacturing in Pharmaceutical Process Development and Manufacturing
- ICH guideline Q13 on continuous manufacturing of drug substances and drug products – Scientific guideline
- Continuous Pharmaceutical Manufacturing: Current trends and future possibilities
- Data Integrity: History, Issues, and Remediation of Issues

Healthcare business analyst with expertise in marketing and business development, and holds an MPharm degree. He specialises in creating and executing communication strategies that make digital health solutions and pharmaceutical technologies clear, accessible, and resonation for their audiences.
link to the author’s linkedin profile



