


Computer System Validation (CSV) in pharma manufacturing is the documented process of proving that a software system consistently does exactly what it is designed to do — within a regulated GxP environment. It provides evidence that digital tools are fit for their intended use, protecting data integrity, patient safety, and regulatory compliance across the entire software lifecycle.
Pharmaceutical manufacturing demands absolute precision. Computer System Validation (CSV) exists to prove digital tools work exactly as intended for patient safety. Research indicates that approximately 25% of FDA enforcement letters include violations related to data validity or fitness-for-purpose evidence. These failures typically stem from poor lifecycle control. Risk-based CSV corrects this to maintain data integrity through the entire lifecycle, and here is how.
Key takeaways:
- Computer System Validation in Pharma prioritizes “fitness for intended use” over technical perfection.
- Data integrity is the primary goal, requiring a lifecycle approach that maintains compliance from design through retirement.
- GAMP 5 prevents “validation fatigue” by focusing rigorous testing on high-risk custom features while streamlining standard tools.
- Early validation is an investment because correcting a system defect after release can cost up to 100 times more than addressing it during the design phase.
- Continuous lifecycle management is critical for avoiding enforcement, as 24% of recent FDA warning letters cite data integrity deficiencies often caused by poor maintenance of the validated state.
What is computer system validation (CSV) in Pharmaceutical Manufacturing?
Computer System Validation meaning in Pharma is having documented evidence that a software application fulfills its intended reliably, and repeatedly. The process validates a system within its specific operating environment and confirms that the system produces accurate results consistently.
Regulators treat unvalidated systems as uncontrolled risks. The FDA mandates this verification to prevent data corruption. Manufacturers must prove their digital infrastructure maintains data integrity. CSV covers the entire software lifecycle. This spans from initial design to retirement. [1]
Expert Takeaway: Validation targets fitness for intended use rather than software perfection. A system might be bug-free yet fail validation if it misses the specific business requirement.
— Kurt In Albon, Principal Pharma Validation Expert | LinkedIn: https://linkedin.com/in/kurtinalbon
What Problem Does CSV Solve in Pharma Manufacturing?
“Fit for intended use” stands as the absolute core of validation. Standard software testing confirms functionality, but CSV confirms context. The process forces technical code to align with strict Good Manufacturing Practice (GMP) requirements. It acts as the vital bridge between IT capabilities and quality assurance mandates.
Failures in this area rarely look like system crashes. They manifest as data integrity gaps. Regulators flag these as inspection findings when audit trails fail, or electronic records become untraceable. The system instantly loses credibility.
A CSV failure is usually a failure of evidence. If the data trail cannot prove the system worked correctly at a specific moment, the product quality becomes suspect.
— Kurt In Albon, Principal Pharma Validation Expert | LinkedIn: https://linkedin.com/in/kurtinalbon
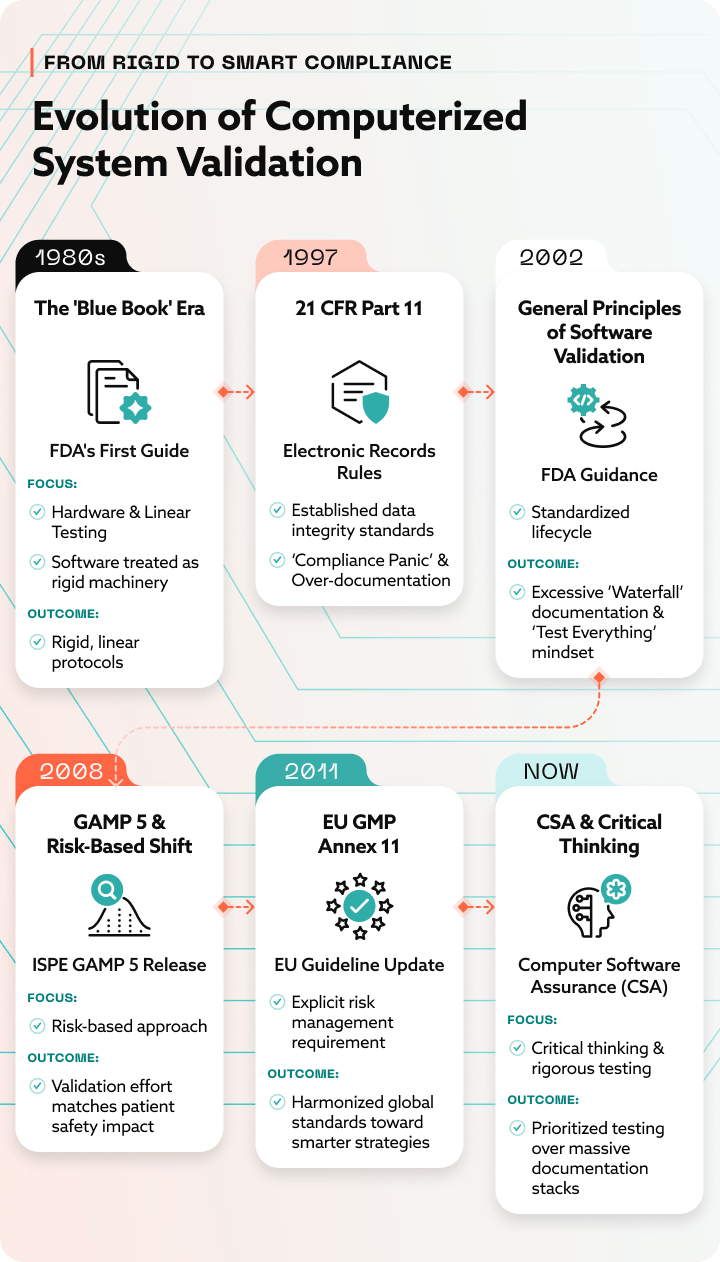
Brief history of CSV in pharma
Which manufacturing systems typically require CSV?
Validation applies to any software directly influencing product quality or data integrity. It encompasses manufacturing execution systems (MES) and laboratory information management systems (LIMS) alongside quality management systems (QMS). Not all systems require the same scrutiny. Here are the categories according to GAMP 5 guidelines:
- Category 1 (Infrastructure): Standard operating systems require only version recording and basic verification.
- Category 3 (Non-Configurable): “Off-the-shelf” tools need validation only for business process alignment.
- Category 4 (Configured): Adaptable systems like LIMS require testing of specific workflow settings.
- Category 5 (Custom): Bespoke software demands full lifecycle validation due to high inherent risk.
Wondering how to know when to use CSV and when CSA? Check out the CSV vs CSA – risk profiles and when each makes sense in our blog article. -.
What Is Risk-Based CSV According to GAMP 5?
“If it wasn’t documented, it didn’t happen.” This old industry maxim ruled validation for decades. GAMP 5 replaces that checkbox mentality with critical thinking. Industry now scales validation effort directly with risk. System complexity and GxP (Good Practice) regulations dictate the testing depth. A standard printer needs verification; a custom algorithm needs stress testing.
“Testing everything” actually introduces compliance risk by diluting focus. FDA analyses reveal that ~95% of cited violations stem from incorrect system design or application, not missing paperwork.Risk-based CSV focuses resources where failure matters most. We validate to ensure safety, not just to fill binders.
What CSV Lifecycle Do Regulators Expect?
Validation often fails because teams treat it as a project and not a process. The Cureus analysis identifies lifecycle gaps as a recurring root cause of non-complian validated status the moment lifecycle controls stop after go-live. Therefore the lifescycle goes in few steps:
|
CSV Lifecycle Phase |
What Happens in This Phase |
Why Regulators Care |
|
Planning & Validation Strategy |
Define the validation roadmap before writing a single test script. Conduct risk assessment to determine the required testing depth. |
Ensures validation effort is proportional to patient and product risk. |
|
Intended Use & User Requirement Specification (URS) |
Document exactly what the system must do within the manufacturing workflow. Establish clear, testable requirements. |
URS becomes the foundation for traceability and all validation testing. |
|
Qualification & Testing (IQ/OQ/PQ) |
Installation Qualification (IQ): verify the system environment. Operational Qualification (OQ): confirm features operate correctly. Performance Qualification (PQ): validate performance under real production conditions. |
Demonstrates the system consistently performs as intended. |
|
Validation Summary & Release |
The Validation Summary Report (VSR) compiles all testing evidence and confirms the system is fit for intended use. |
Provides the primary documented defense during regulatory inspections. |
|
Maintenance of Validated State |
Apply strict change control for patches, upgrades, or configuration changes. Maintain documentation and revalidate when necessary. |
Prevents loss of validated status after go-live. |
|
System Retirement |
Execute a documented decommissioning plan. Migrate or archive records to maintain accessibility for required retention periods. |
Protects long-term data integrity and regulatory traceability. |
Expert Contributor: Kurt In Albon, Principal Pharma Validation Expert
Intended use, user requirements, and process understanding
Effective CSV starts with the manufacturing process: “You cannot validate what you do not understand.” You should define exactly which GMP decisions the system supports before writing a single requirement. Unclear intended use creates vague protocols that fail to detect critical risks during production operations.
In the Intelligent Pharma Manufacturing scenario, deep process understanding allows the team to automate batch-release logic safely. This precision reduces release cycle times and eliminates manual verification errors. Weak process definition, conversely, leads to validation gaps where the software passes technical testing but fails to stop a non-compliant product. [3]
Why Is Data Integrity the Core Outcome of CSV?
Regulators rarely cite CSV mechanics in isolation. They target the data integrity failures that validation was meant to prevent. Systems need to be validated to ensure that every record remains attributable and accurate throughout its retention period.
ALCOA+ principles guide this defensive strategy. Specific controls like locked audit trails and strict access rights should be implemented to protect the data’s truth. Effective CSV verifies that electronic signatures and change controls function correctly to prevent unauthorized modifications that could hide quality issues. [4]
What Are the 8 Steps to Implement and Validate a GxP Computerized System?
A structured lifecycle ensures compliance from the start. Studies show that fixing a defect after release costs up to 100 times more than addressing it during the design phase. [5]
Phase 1 – Process and system understanding
The process must be mapped to visualize the exact workflow. This step identifies where the system interacts with human operators. Defining system boundaries locks down the project scope early.
Phase 2 – Intended use and system classification
Intended use defines the specific business problem the system solves. This definition drives the GAMP 5 category – a standard tool requires minimal effort, but a custom engine demands full structural testing.
Phase 3 – User requirements specification
The URS and functional requirements specification (FRS) serve as the project’s constitution. Process-driven requirements must take precedence over technical features. Research indicates that approximately 70% of product defects originate in the planning and requirements phase, making this step critical. [6]
A resource that can help you write a URS: How to Write a URS: Expert Guidance
Phase 4 – Risk assessment
A risk-based approach focuses testing effort on specific impact areas. The team identifies risks to patient safety or data integrity. High-risk functions receive rigorous challenge testing. Low-risk features undergo simple verification.
Phase 5 – System configuration and implementation
Strict distinction between configuration (standard settings) and customization (new code) is vital. Traceability links every build decision back to a specific user requirement. Controlled implementation ensures the system matches the approved design before formal testing begins.
Phase 6 – Verification and testing
Testing follows the “V-Model” progression. Testing alone detects only about small amount of defects, which reinforces the need for robust design controls in earlier phases. [7]
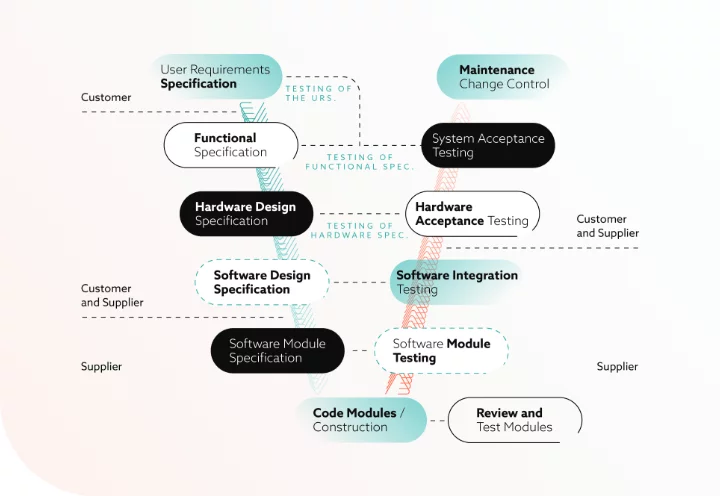
Phase 7 – Validation reporting and release
The VSR consolidates all testing evidence. It provides a clear conclusion on the system’s fitness for use. Quality Assurance reviews this report to ensure all deviations are resolved. Formal GxP release authorizes the system for production use only after this approval is signed.
Phase 8 – Operation, change control, and continuous validation
Post-go-live, every update is governed by a standard operating procedure (SOP) and change control to preserve the validated state. This is crucial, as 53.5% of FDA warning letters in recent years have cited data integrity deficiencies often linked to poor lifecycle management. [8]
Common CSV pitfalls in pharma manufacturing
- Unclear intended use: Systems are often validated against technical specs rather than business needs. This disconnect leads to “not fit for purpose” findings because the system passes testing but fails to control the actual GMP process risk.
- Over-documentation: Teams often prioritize volume over value. This “wall of paper” obscures real risks, though modern CSA frameworks now encourage critical thinking to reduce this burden.
- Weak change control: A system is only validated until the first unapproved patch. Poor change management leads to a “broken validated state,” where undocumented updates silently drift the system out of compliance.
- Poor ownership: When IT owns the server but Quality owns the data, accountability gaps emerge. This fragmented ownership leads to inconsistent execution. [1]
How Can Digital Validation Platforms Support CSV?
BGO Software delivers end-to-end Computer Software Validation consulting and custom software development tailored specifically to pharmaceutical and life sciences environments. Whether you need expert guidance navigating a complex validation project or a purpose-built validated system designed from the ground up, our team brings deep regulatory knowledge and hands-on GxP experience to every engagement.
Supporting the full validation lifecycle from risk assessments and URS authorship through to IQ/OQ/PQ execution and validation summary reports and ensuring your systems are compliant, audit-ready, and built on a defensible documentation trail. For organisations requiring custom software development, BGO builds solutions with validation built in from day one, not retrofitted after go-live.
The custom platforms that now take weeks and not years to built and fully validate can integrate seamlessly with existing enterprise systems such as QMS, MES, ERP, LIMS, and DMS to eliminating siloed data and delivering a validated data layer that provides real-time operational insight.
By working with a partner who understands both the regulatory landscape and the technical complexity of regulated software, your teams can accelerate validation timelines, maintain data integrity by design, and stay inspection-ready — without adding unnecessary complexity to your processes.

Healthcare business analyst with expertise in marketing and business development, and holds an MPharm degree. He specialises in creating and executing communication strategies that make digital health solutions and pharmaceutical technologies clear, accessible, and resonation for their audiences.
link to the author’s linkedin profile



